

Contact order
Encyclopedia
The contact order of a protein
is a measure of the locality of the inter-amino acid
contacts in the protein's native state
tertiary structure
. It is calculated as the average sequence distance between residues that form native contact
s in the folded protein divided by the total length of the protein. Higher contact orders indicate longer folding times
, and low contact order has been suggested as a predictor of potential downhill folding
, or protein folding that occurs without a free energy
barrier. This effect is thought to be due to the lower loss of conformational entropy
associated with the formation of local as opposed to nonlocal contacts.
Contact order (CO) is formally defined as: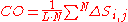
where N is the total number of contacts, ΔSi,j is the sequence separation, in residues, between contacting residues i and j, and L is the total number of residues in the protein. The value of contact order typically ranges from 5% to 25% for single-domain proteins, with lower contact order belonging to mainly helical proteins, and higher contact order belonging to proteins with a high beta-sheet content.
Protein structure prediction
methods are more accurate in predicting the structures of proteins with low contact orders. This may be partly because low contact order proteins tend to be small, but is likely to be explained by the smaller number of possible long-range residue-residue interactions to be considered during global optimization
procedures that minimize an energy function. Even successful structure prediction methods such as the Rosetta
method overproduce low-contact-order structure predictions compared to the distributions observed in experimentally determined protein structures.
The percentage of the natively folded contact order can also be used as a measure of the "nativeness" of folding transition state
s. Phi value analysis
in concert with molecular dynamics
has produced transition-state models whose contact order is close to that of the folded state in proteins that are small and fast-folding. Further, contact orders in transition states as well as those in native states are highly correlated with overall folding time.
In addition to their role in structure prediction, contact orders can themselves be predicted based on a sequence alignment
, which can be useful in classifying the fold of a novel sequence with some degree of homology
to known sequences.
Protein
Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form, facilitating a biological function. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of...
is a measure of the locality of the inter-amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
contacts in the protein's native state
Native state
In biochemistry, the native state of a protein is its operative or functional form. While all protein molecules begin as simple unbranched chains of amino acids, once completed they assume highly specific three-dimensional shapes; that ultimate shape, known as tertiary structure, is the folded...
tertiary structure
Tertiary structure
In biochemistry and molecular biology, the tertiary structure of a protein or any other macromolecule is its three-dimensional structure, as defined by the atomic coordinates.-Relationship to primary structure:...
. It is calculated as the average sequence distance between residues that form native contact
Native contact
In protein folding, a native contact is a contact between the side chains of two amino acids that are not neighboring in the amino acid sequence but are spatially close in the protein's native state tertiary structure...
s in the folded protein divided by the total length of the protein. Higher contact orders indicate longer folding times
Protein folding
Protein folding is the process by which a protein structure assumes its functional shape or conformation. It is the physical process by which a polypeptide folds into its characteristic and functional three-dimensional structure from random coil....
, and low contact order has been suggested as a predictor of potential downhill folding
Downhill folding
Downhill folding is a process in which a protein folds without encountering any significant macroscopic free energy barrier. It is a key prediction of the folding funnel hypothesis of the energy landscape theory of proteins.-Overview:...
, or protein folding that occurs without a free energy
Thermodynamic free energy
The thermodynamic free energy is the amount of work that a thermodynamic system can perform. The concept is useful in the thermodynamics of chemical or thermal processes in engineering and science. The free energy is the internal energy of a system less the amount of energy that cannot be used to...
barrier. This effect is thought to be due to the lower loss of conformational entropy
Conformational entropy
Conformational entropy is the entropy associated with the physical arrangement of a polymer chain that assumes a compact or globular state in solution. The concept is most commonly applied to biological macromolecules such as proteins and RNA, but can also be used for polysaccharides and other...
associated with the formation of local as opposed to nonlocal contacts.
Contact order (CO) is formally defined as:
where N is the total number of contacts, ΔSi,j is the sequence separation, in residues, between contacting residues i and j, and L is the total number of residues in the protein. The value of contact order typically ranges from 5% to 25% for single-domain proteins, with lower contact order belonging to mainly helical proteins, and higher contact order belonging to proteins with a high beta-sheet content.
Protein structure prediction
Protein structure prediction
Protein structure prediction is the prediction of the three-dimensional structure of a protein from its amino acid sequence — that is, the prediction of its secondary, tertiary, and quaternary structure from its primary structure. Structure prediction is fundamentally different from the inverse...
methods are more accurate in predicting the structures of proteins with low contact orders. This may be partly because low contact order proteins tend to be small, but is likely to be explained by the smaller number of possible long-range residue-residue interactions to be considered during global optimization
Global optimization
Global optimization is a branch of applied mathematics and numerical analysis that deals with the optimization of a function or a set of functions to some criteria.- General :The most common form is the minimization of one real-valued function...
procedures that minimize an energy function. Even successful structure prediction methods such as the Rosetta
Rosetta@home
Rosetta@home is a distributed computing project for protein structure prediction on the Berkeley Open Infrastructure for Network Computing platform, run by the Baker laboratory at the University of Washington...
method overproduce low-contact-order structure predictions compared to the distributions observed in experimentally determined protein structures.
The percentage of the natively folded contact order can also be used as a measure of the "nativeness" of folding transition state
Transition state
The transition state of a chemical reaction is a particular configuration along the reaction coordinate. It is defined as the state corresponding to the highest energy along this reaction coordinate. At this point, assuming a perfectly irreversible reaction, colliding reactant molecules will always...
s. Phi value analysis
Phi value analysis
Phi value analysis is an experimental protein engineering method used to study the structure of the folding transition state in small protein domains that fold in a two-state manner. Since the folding transition state is by definition a transient and partially unstructured state, its structure is...
in concert with molecular dynamics
Molecular dynamics
Molecular dynamics is a computer simulation of physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a period of time, giving a view of the motion of the atoms...
has produced transition-state models whose contact order is close to that of the folded state in proteins that are small and fast-folding. Further, contact orders in transition states as well as those in native states are highly correlated with overall folding time.
In addition to their role in structure prediction, contact orders can themselves be predicted based on a sequence alignment
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
, which can be useful in classifying the fold of a novel sequence with some degree of homology
Homology (biology)
Homology forms the basis of organization for comparative biology. In 1843, Richard Owen defined homology as "the same organ in different animals under every variety of form and function". Organs as different as a bat's wing, a seal's flipper, a cat's paw and a human hand have a common underlying...
to known sequences.