

FASTQ format
Encyclopedia
FASTQ format is a text-based format
for storing both a biological sequence (usually nucleotide sequence) and its corresponding quality scores. Both the sequence letter and quality score are encoded with a single ASCII
character for brevity. It was originally developed at the Wellcome Trust Sanger Institute to bundle a FASTA
sequence and its quality data, but has recently become the de facto standard for storing the output of high throughput sequencing instruments such as the Illumina Genome Analyzer
.
title line). Line 2 is the raw sequence letters. Line 3 begins with a '+' character and is optionally followed by the same sequence identifier (and any description) again. Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
A minimal FASTQ file might look like this:
The original Sanger FASTQ files also allowed the sequence and quality strings to be wrapped (split over multiple lines), but this is generally discouraged as it can make parsing complicated due to the unfortunate choice of "@" and "+" as markers (these characters can also occur in the quality string).
Versions of the Illumina pipeline since 1.4 appear to use #NNNNNN instead of #0 for the multiplex ID, where NNNNNN is the sequence of the multiplex tag.
With Casava 1.8 the format of the '@' line has changed:
/EBI
Sequence Read Archive
often include a description, e.g.
In this example there is an NCBI-assigned identifier, and the description holds the original identifier from Solexa/Illumina (as described above) plus the read length.
Also note that the NCBI have converted this FASTQ data from the original Solexa/Illumina encoding to the Sanger standard (see encodings below).
:

The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used a different mapping, encoding the odds
p/(1-p) instead of the probability p:
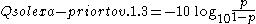
Although both mappings are asymptotically identical at higher quality values, they differ at lower quality levels (i.e., approximately p > 0.05, or equivalently, Q < 13).
At times there has been disagreement about which mapping Illumina actually uses. The user guide (Appendix B, page 122) for version 1.4 of the Illumina pipeline states that: "The scores are defined as Q=10*log10(p/(1-p)) [sic], where p is the probability of a base call corresponding to the base in question". In retrospect, this entry in the manual appears to have been an error. The user guide (What's New, page 5) for version 1.5 of the Illumina pipeline lists this description instead: "Important Changes in Pipeline v1.3 [sic]. The quality scoring scheme has changed to the Phred [i.e., Sanger] scoring scheme, encoded as an ASCII character by adding 64 to the Phred value. A Phred score of a base is: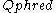 =-10
=-10  (e), where e is the estimated probability of a base being wrong.
(e), where e is the estimated probability of a base being wrong.
An alternative interpretation of this ASCII encoding has been proposed. Also, in Illumina runs using PhiX controls, the character 'B' was observed to represent an "unknown quality score". The error rate of 'B' reads was roughly 3 phred scores lower the mean observed score of a given run.
For raw reads, the range of scores will depend on the technology and the base caller used, but will typically be up to 40. Recent Illumina chemistry changes have resulted in reported quality scores of 41, which has broken various scripts and tools expecting an upper bound of 40. For aligned sequences and consensuses higher scores are common.
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS.....................................................
..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX......................
...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII......................
.................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ......................
LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL....................................................
!"#$%&'*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
| | | | | |
33 59 64 73 104 126
S - Sanger Phred+33, raw reads typically (0, 40)
X - Solexa Solexa+64, raw reads typically (-5, 40)
I - Illumina 1.3+ Phred+64, raw reads typically (0, 40)
J - Illumina 1.5+ Phred+64, raw reads typically (3, 40)
with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold)
(Note: See discussion above).
L - Illumina 1.8+ Phred+33, raw reads typically (0, 41)
File format
A file format is a particular way that information is encoded for storage in a computer file.Since a disk drive, or indeed any computer storage, can store only bits, the computer must have some way of converting information to 0s and 1s and vice-versa. There are different kinds of formats for...
for storing both a biological sequence (usually nucleotide sequence) and its corresponding quality scores. Both the sequence letter and quality score are encoded with a single ASCII
ASCII
The American Standard Code for Information Interchange is a character-encoding scheme based on the ordering of the English alphabet. ASCII codes represent text in computers, communications equipment, and other devices that use text...
character for brevity. It was originally developed at the Wellcome Trust Sanger Institute to bundle a FASTA
FASTA format
In bioinformatics, FASTA format is a text-based format for representing either nucleotide sequences or peptide sequences, in which nucleotides or amino acids are represented using single-letter codes. The format also allows for sequence names and comments to precede the sequences...
sequence and its quality data, but has recently become the de facto standard for storing the output of high throughput sequencing instruments such as the Illumina Genome Analyzer
Illumina (company)
Illumina, Inc. is a company incorporated in April 1998 that develops, manufactures and markets integrated systems for the analysis of genetic variation and biological function. Using its technologies, the company provides a line of products and services that serve the sequencing, genotyping and...
.
Format
A FASTQ file normally uses four lines per sequence. Line 1 begins with a '@' character and is followed by a sequence identifier and an optional description (like a FASTAFASTA format
In bioinformatics, FASTA format is a text-based format for representing either nucleotide sequences or peptide sequences, in which nucleotides or amino acids are represented using single-letter codes. The format also allows for sequence names and comments to precede the sequences...
title line). Line 2 is the raw sequence letters. Line 3 begins with a '+' character and is optionally followed by the same sequence identifier (and any description) again. Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
A minimal FASTQ file might look like this:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!*((((***+))%%%++)(%%%%).1***-+*))**55CCF>>>>>>CCCCCCC65
The original Sanger FASTQ files also allowed the sequence and quality strings to be wrapped (split over multiple lines), but this is generally discouraged as it can make parsing complicated due to the unfortunate choice of "@" and "+" as markers (these characters can also occur in the quality string).
Illumina sequence identifiers
Sequences from the Illumina software use a systematic identifier:
@HWUSI-EAS100R:6:73:941:1973#0/1
| HWUSI-EAS100R | the unique instrument name |
|---|---|
| 6 | flowcell lane |
| 73 | tile number within the flowcell lane |
| 941 | 'x'-coordinate of the cluster within the tile |
| 1973 | 'y'-coordinate of the cluster within the tile |
| #0 | index number for a multiplexed sample (0 for no indexing) |
| /1 | the member of a pair, /1 or /2 (paired-end or mate-pair reads only) |
Versions of the Illumina pipeline since 1.4 appear to use #NNNNNN instead of #0 for the multiplex ID, where NNNNNN is the sequence of the multiplex tag.
With Casava 1.8 the format of the '@' line has changed:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
| EAS139 | the unique instrument name |
|---|---|
| 136 | the run id |
| FC706VJ | the flowcell id |
| 2 | flowcell lane |
| 2104 | tile number within the flowcell lane |
| 15343 | 'x'-coordinate of the cluster within the tile |
| 197393 | 'y'-coordinate of the cluster within the tile |
| 1 | the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| Y | Y if the read fails filter (read is bad), N otherwise |
| 18 | 0 when none of the control bits are on, otherwise it is an even number |
| ATCACG | index sequence |
NCBI Sequence Read Archive
FASTQ files from the NCBINational Center for Biotechnology Information
The National Center for Biotechnology Information is part of the United States National Library of Medicine , a branch of the National Institutes of Health. The NCBI is located in Bethesda, Maryland and was founded in 1988 through legislation sponsored by Senator Claude Pepper...
/EBI
European Bioinformatics Institute
The European Bioinformatics Institute is a centre for research and services in bioinformatics, and is part of European Molecular Biology Laboratory...
Sequence Read Archive
Sequence Read Archive
The Sequence Read Archive or Short Read Archive is an important bioinformatics database hosted at the European Bioinformatics Institute. It provides a public repository for the 'short reads' generated by second generation sequencing technologies....
often include a description, e.g.
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
In this example there is an NCBI-assigned identifier, and the description holds the original identifier from Solexa/Illumina (as described above) plus the read length.
Also note that the NCBI have converted this FASTQ data from the original Solexa/Illumina encoding to the Sanger standard (see encodings below).
Quality
A quality value Q is an integer mapping of p (i.e., the probability that the corresponding base call is incorrect). Two different equations have been in use. The first is the standard Sanger variant to assess reliability of a base call, otherwise known as Phred quality scorePhred quality score
Phred quality scores were originally developed by the program Phred to help in the automation of DNA sequencing in the Human Genome Project. Phred quality scores are assigned to each base call in automated sequencer traces...
:
The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used a different mapping, encoding the odds
Odds
The odds in favor of an event or a proposition are expressed as the ratio of a pair of integers, which is the ratio of the probability that an event will happen to the probability that it will not happen...
p/(1-p) instead of the probability p:
Although both mappings are asymptotically identical at higher quality values, they differ at lower quality levels (i.e., approximately p > 0.05, or equivalently, Q < 13).
At times there has been disagreement about which mapping Illumina actually uses. The user guide (Appendix B, page 122) for version 1.4 of the Illumina pipeline states that: "The scores are defined as Q=10*log10(p/(1-p)) [sic], where p is the probability of a base call corresponding to the base in question". In retrospect, this entry in the manual appears to have been an error. The user guide (What's New, page 5) for version 1.5 of the Illumina pipeline lists this description instead: "Important Changes in Pipeline v1.3 [sic]. The quality scoring scheme has changed to the Phred [i.e., Sanger] scoring scheme, encoded as an ASCII character by adding 64 to the Phred value. A Phred score of a base is:
=-10 (e), where e is the estimated probability of a base being wrong.Encoding
- Sanger format can encode a Phred quality scorePhred quality scorePhred quality scores were originally developed by the program Phred to help in the automation of DNA sequencing in the Human Genome Project. Phred quality scores are assigned to each base call in automated sequencer traces...
from 0 to 93 using ASCIIASCIIThe American Standard Code for Information Interchange is a character-encoding scheme based on the ordering of the English alphabet. ASCII codes represent text in computers, communications equipment, and other devices that use text...
33 to 126 (although in raw read data the Phred quality score rarely exceeds 60, higher scores are possible in assemblies or read maps). Also used in SAM format. Coming to the end of February 2011, Illumina's newest version (1.8) of their pipeline CASAVA will produce directly fastq in Sanger format, according to the announcement on seqanswers.com forum. - Solexa/Illumina 1.0 format can encode a Solexa/Illumina quality score from -5 to 62 using ASCIIASCIIThe American Standard Code for Information Interchange is a character-encoding scheme based on the ordering of the English alphabet. ASCII codes represent text in computers, communications equipment, and other devices that use text...
59 to 126 (although in raw read data Solexa scores from -5 to 40 only are expected) - Illumina 1.3+ format can encode a Phred quality scorePhred quality scorePhred quality scores were originally developed by the program Phred to help in the automation of DNA sequencing in the Human Genome Project. Phred quality scores are assigned to each base call in automated sequencer traces...
from 0 to 62 using ASCIIASCIIThe American Standard Code for Information Interchange is a character-encoding scheme based on the ordering of the English alphabet. ASCII codes represent text in computers, communications equipment, and other devices that use text...
64 to 126 (although in raw read data Phred scores from 0 to 40 only are expected). - The Phred scores 0 to 2 in Illumina 1.5+ have a slightly different meaning. The values 0 and 1 are no longer used and the value 2, encoded by ASCII 66 "B", is used also at the end of reads as a Read Segment Quality Control Indicator . The Illumina manual (page 30) states the following: If a read ends with a segment of mostly low quality (Q15 or below), then all of the quality values in the segment are replaced with a value of 2 (encoded as the letter B in Illumina's text-based encoding of quality scores)... This Q2 indicator does not predict a specific error rate, but rather indicates that a specific final portion of the read should not be used in further analyses. Also, the quality score encoded as "B" letter may occur internally within reads at least as late as pipeline version 1.6, as shown in the following example:
@HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1
TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT
+HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1
efcfffffcfeefffcffffffddf`feed]`]_Ba_^__[YBBBBBBBBBBRTT\]][]dddd`ddd^dddadd^BBBBBBBBBBBBBBBBBBBBBBBB
An alternative interpretation of this ASCII encoding has been proposed. Also, in Illumina runs using PhiX controls, the character 'B' was observed to represent an "unknown quality score". The error rate of 'B' reads was roughly 3 phred scores lower the mean observed score of a given run.
For raw reads, the range of scores will depend on the technology and the base caller used, but will typically be up to 40. Recent Illumina chemistry changes have resulted in reported quality scores of 41, which has broken various scripts and tools expecting an upper bound of 40. For aligned sequences and consensuses higher scores are common.
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS.....................................................
..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX......................
...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII......................
.................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ......................
LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL....................................................
!"#$%&'*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
| | | | | |
33 59 64 73 104 126
S - Sanger Phred+33, raw reads typically (0, 40)
X - Solexa Solexa+64, raw reads typically (-5, 40)
I - Illumina 1.3+ Phred+64, raw reads typically (0, 40)
J - Illumina 1.5+ Phred+64, raw reads typically (3, 40)
with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold)
(Note: See discussion above).
L - Illumina 1.8+ Phred+33, raw reads typically (0, 41)
Color space
For SOLiD data, the sequence is in color space, except the first position. The quality values are those of the Sanger format. Alignment tools differ in their preferred version of the quality values: some include a quality score (set to 0, i.e. '!') for the leading nucleotide, others do not. The sequence read archive includes this quality score.File extension
There is no standard file extension for a FASTQ file, but .fq, .fastq, and .txt are commonly used.Format converters
- BiopythonBioPythonThe Biopython Project is an international association of developers of freely available Python tools for computational molecular biology, as well as bioinformatics.-References:*refer to the Biopython website for other , and a list of over one hundred ....
version 1.51 onwards (interconverts Sanger, Solexa and Illumina 1.3+) - EMBOSSEMBOSSEMBOSS is an acronym for European Molecular Biology Open Software Suite. EMBOSS is a free Open Source software analysis package specially developed for the needs of the molecular biology and bioinformatics user community...
version 6.1.0 patch 1 onwards (interconverts Sanger, Solexa and Illumina 1.3+) - BioPerlBioPerlBioPerl is a collection of Perl modules that facilitate the development of Perl scripts for bioinformatics applications. It has played an integral role in the Human Genome Project....
version 1.6.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+) - BioRubyBioRubyBioRuby is a package of Open Source Ruby code, with classes for DNA and protein sequence analysis, alignment, database parsing, and other Bioinformatics tools. Recently, tools for structural biology have been added.-External links:* ,*...
version 1.4.0 onwards (interconverts Sanger, Solexa and Illumina 1.3+) - MAQ can convert from Solexa to Sanger (use this patch to support Illumina 1.3+ files).
See also
- FASTA formatFASTA formatIn bioinformatics, FASTA format is a text-based format for representing either nucleotide sequences or peptide sequences, in which nucleotides or amino acids are represented using single-letter codes. The format also allows for sequence names and comments to precede the sequences...
- List of file formats for molecular biology
External links
- MAQ webpage discussing FASTQ variants
- Galaxy fastq tools
- Fastx toolkit collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing
- Fastqc quality control tool for high throughput sequence data
- PRINSEQ can be used for QC and to filter, reformat, or trim sequence data (web-based and command line versions)