
Glycine encephalopathy
Encyclopedia
Glycine encephalopathy is a rare autosomal recessive disorder of glycine
metabolism. After phenylketonuria
, glycine encephalopathy is the second most common disorder of amino acid
metabolism. The disease is caused by defects in the glycine cleavage system
, an enzyme
responsible for glycine catabolism. There are several forms of the disease, with varying severity of symptoms and time of onset. The symptoms are exclusively neurological in nature, and clinically this disorder is characterized by abnormally high levels of the amino acid glycine
in bodily fluids and tissues, especially the cerebral spinal fluid.
Glycine encephalopathy is sometimes referred to as "nonketotic hyperglycinemia" (NKH), as a reference to the biochemical findings seen in patients with the disorder, and to distinguish it from the disorders that cause "ketotic hyperglycinemia" (seen in propionic acidemia
and several other inherited metabolic disorders). To avoid confusion, the term "glycine encephalopathy" is often used, as this term more accurately describes the clinical symptoms of the disorder.
in the brain, acting as an inhibitor in the spinal cord and brain stem, while having excitatory in the cortex of the brain. Glycine is metabolized to eventual end products of ammonia
and carbon dioxide
through the glycine cleavage system
(GCS), an enzyme complex
made up of four protein subunits. Defects in these subunits can cause glycine encephalopathy, although some causes of the disease are still unknown. The GCS has its highest activity levels in liver, brain and placental tissue. One of its main functions is to keep glycine levels low in the brain. Defects in GCS cause an increase of glycine concentration in plasma
and cerebrospinal fluid
. The exact pathophysiology of the disorder is not known, but it is considered likely that buildup of glycine in the brain is responsible for the symptoms.
All types on glycine encephalopathy show elevated levels of glycine in the plasma, and cerebral spinal fluid. Glycine concentrations in the cerebrospinal fluid are typically more markedly elevated than in plasma, leading to a corresponding elevation in the ratio of glycine concentrations in the cerebral spinal fluid to that in the plasma. The ratio can also be slightly elevated if the patient is receiving valproic acid
.
Glycine encephalopathy (nonketotic hyperglycinemia) should not be confused with other metabolic disorders that can produce elevated glycine. For example, certain inherited 'organic acidurias' (aka 'organic acidemias') can produce elevated glycine in plasma and urine, although the disorders are not caused by defects in the glycine cleavage system. Glycine encephalopathy is unique in the fact that levels of glycine are disproportionately elevated in CSF (in addition to blood and urine), whereas CSF glycine levels are normal or near-normal in patients with inherited organic acidurias.
and ammonia
. The glycine cleavage system
, which is responsible for glycine metabolism in the mitochondrion
is made up of four protein subunits, the P-protein, H-protein, T-protein and L-protein.
, hypotonia
, seizures and brain malformations.
With the classical, or neonatal presentation of glycine encephalopathy, the infant is born after an unremarkable pregnancy, but presents with lethargy, hypotonia
, seizures and myoclonic jerks, which can progress to apnea
requiring artificial ventilation, and often death. Apneic patients can regain spontaneous respiration in their second to third week of life. After recovery from the initial episode, patients have intractable seizures and profound mental retardation
, remaining developmentally delayed. Some mothers comment retrospectively that they noticed rhythmic hiccuping during pregnancy. Patients with the infantile form of glycine encephalopathy do not show lethargy and coma in the neonatal period, but often have a history of hypotonia. They often have seizures, which can range in severity and responsiveness to treatment, and are typically developmentally delayed. Glycine encephalopathy can also present as a milder form with episodic seizures, ataxia
, movement disorders, and gaze palsy during febrile illness. These patients are also developmentally delayed, to varying degrees. In the later onset form, patients typically have normal intellectual function, but present with spastic diplegia
and optic atrophy.
Transient neonatal hyperglycinemia has been described in a very small number of cases. Initially, these patients present with the same symptoms and laboratory results that are seen in the classical presentation. The levels of glycine in plasma and cerebrospinal fluid normalize within eight weeks, and in five of six cases there were no neurological issues at follow-up times up to thirteen years. A single patient was severely retarded at nine months. An immature glycine cleavage system in the brain and liver is suspected as the cause of transient neonatal hyperglicinemia.
 Glycine encephalopathy has an estimated incidence of 1 in 60,000, making it the second most common disorder of amino acid metabolism, after phenylketonuria
Glycine encephalopathy has an estimated incidence of 1 in 60,000, making it the second most common disorder of amino acid metabolism, after phenylketonuria
. It is caused by a defect in the glycine cleavage system
(GCS), which is made up of four protein subunits. Each of these four subunits is encoded by a separate gene
. Defects in three of these four genes have been linked to glycine encephalopathy.
There is a fourth unit in the complex, dihydrolipoamide dehydrogenase
or GCSL. However, to date there have been no mutations in GCSL found to be associated with glycine encephalopathy.
A small percentage of affected individuals do not have a detectable mutation in any of the three genes (listed above) but still have defective glycine-cleavage enzymatic activity. It is thought that these patients may have mutations in the genes encoding one of the cofactors associated with the GCS complex.
Defects in the GSC proteins can prevent the complex from functioning properly or can prevent the GCS complex from forming entirely. When the complex is unable to break down glycine properly, this causes excess glycine to build up to toxic levels in the body's organs and tissues. Damage caused by harmful levels of glycine in the brain and cerebrospinal fluid
is responsible for the characteristic seizures, breathing difficulties, movement disorders, and mental retardation.
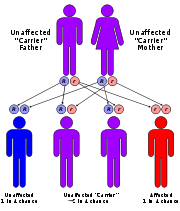
This disorder is inherited in an autosomal recessive pattern. The term "autosomal" signifies that the gene associated with the disorder is located on an autosome
. In an autosomal recessive inheritance pattern, two defective copies of the gene (one inherited from each parent) are required in order for a child to be born with the disorder. Therefore, each parent of an individual with an autosomal recessive disorder has at least one defective copy of the gene. With autosomal recessive disorders, individuals with only one copy of a defective gene (heterozygotes) are considered "carriers" for the disorder. Carriers usually do not show signs or symptoms of the disorder.
Glycine
Glycine is an organic compound with the formula NH2CH2COOH. Having a hydrogen substituent as its 'side chain', glycine is the smallest of the 20 amino acids commonly found in proteins. Its codons are GGU, GGC, GGA, GGG cf. the genetic code.Glycine is a colourless, sweet-tasting crystalline solid...
metabolism. After phenylketonuria
Phenylketonuria
Phenylketonuria is an autosomal recessive metabolic genetic disorder characterized by a mutation in the gene for the hepatic enzyme phenylalanine hydroxylase , rendering it nonfunctional. This enzyme is necessary to metabolize the amino acid phenylalanine to the amino acid tyrosine...
, glycine encephalopathy is the second most common disorder of amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
metabolism. The disease is caused by defects in the glycine cleavage system
Glycine cleavage system
The glycine cleavage system is also known as the glycine decarboxylase complex or GCS. The system is a series of enzymes that are triggered in response to high concentrations of the amino acid glycine. The glycine cleavage system is composed of four proteins: the T-protein, P-protein, L-protein,...
, an enzyme
Enzyme
Enzymes are proteins that catalyze chemical reactions. In enzymatic reactions, the molecules at the beginning of the process, called substrates, are converted into different molecules, called products. Almost all chemical reactions in a biological cell need enzymes in order to occur at rates...
responsible for glycine catabolism. There are several forms of the disease, with varying severity of symptoms and time of onset. The symptoms are exclusively neurological in nature, and clinically this disorder is characterized by abnormally high levels of the amino acid glycine
Glycine
Glycine is an organic compound with the formula NH2CH2COOH. Having a hydrogen substituent as its 'side chain', glycine is the smallest of the 20 amino acids commonly found in proteins. Its codons are GGU, GGC, GGA, GGG cf. the genetic code.Glycine is a colourless, sweet-tasting crystalline solid...
in bodily fluids and tissues, especially the cerebral spinal fluid.
Glycine encephalopathy is sometimes referred to as "nonketotic hyperglycinemia" (NKH), as a reference to the biochemical findings seen in patients with the disorder, and to distinguish it from the disorders that cause "ketotic hyperglycinemia" (seen in propionic acidemia
Propionic acidemia
Propionic acidemia, also known as propionic aciduria, propionyl-CoA carboxylase deficiency and ketotic glycinemia, is an autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia....
and several other inherited metabolic disorders). To avoid confusion, the term "glycine encephalopathy" is often used, as this term more accurately describes the clinical symptoms of the disorder.
Pathophysiology
Glycine is the simplest amino acid, having no stereoisomers. It can act as a neurotransmitterNeurotransmitter
Neurotransmitters are endogenous chemicals that transmit signals from a neuron to a target cell across a synapse. Neurotransmitters are packaged into synaptic vesicles clustered beneath the membrane on the presynaptic side of a synapse, and are released into the synaptic cleft, where they bind to...
in the brain, acting as an inhibitor in the spinal cord and brain stem, while having excitatory in the cortex of the brain. Glycine is metabolized to eventual end products of ammonia
Ammonia
Ammonia is a compound of nitrogen and hydrogen with the formula . It is a colourless gas with a characteristic pungent odour. Ammonia contributes significantly to the nutritional needs of terrestrial organisms by serving as a precursor to food and fertilizers. Ammonia, either directly or...
and carbon dioxide
Carbon dioxide
Carbon dioxide is a naturally occurring chemical compound composed of two oxygen atoms covalently bonded to a single carbon atom...
through the glycine cleavage system
Glycine cleavage system
The glycine cleavage system is also known as the glycine decarboxylase complex or GCS. The system is a series of enzymes that are triggered in response to high concentrations of the amino acid glycine. The glycine cleavage system is composed of four proteins: the T-protein, P-protein, L-protein,...
(GCS), an enzyme complex
Protein complex
A multiprotein complex is a group of two or more associated polypeptide chains. If the different polypeptide chains contain different protein domain, the resulting multiprotein complex can have multiple catalytic functions...
made up of four protein subunits. Defects in these subunits can cause glycine encephalopathy, although some causes of the disease are still unknown. The GCS has its highest activity levels in liver, brain and placental tissue. One of its main functions is to keep glycine levels low in the brain. Defects in GCS cause an increase of glycine concentration in plasma
Plasma
Plasma may refer to:* Blood plasma, the yellow-colored liquid component of blood, in which blood cells are suspended* Plasma , an ionized state of matter similar to a gas...
and cerebrospinal fluid
Cerebrospinal fluid
Cerebrospinal fluid , Liquor cerebrospinalis, is a clear, colorless, bodily fluid, that occupies the subarachnoid space and the ventricular system around and inside the brain and spinal cord...
. The exact pathophysiology of the disorder is not known, but it is considered likely that buildup of glycine in the brain is responsible for the symptoms.
All types on glycine encephalopathy show elevated levels of glycine in the plasma, and cerebral spinal fluid. Glycine concentrations in the cerebrospinal fluid are typically more markedly elevated than in plasma, leading to a corresponding elevation in the ratio of glycine concentrations in the cerebral spinal fluid to that in the plasma. The ratio can also be slightly elevated if the patient is receiving valproic acid
Valproic acid
Valproic acid is a chemical compound that has found clinical use as an anticonvulsant and mood-stabilizing drug, primarily in the treatment of epilepsy, bipolar disorder, and, less commonly, major depression. It is also used to treat migraine headaches and schizophrenia...
.
Glycine encephalopathy (nonketotic hyperglycinemia) should not be confused with other metabolic disorders that can produce elevated glycine. For example, certain inherited 'organic acidurias' (aka 'organic acidemias') can produce elevated glycine in plasma and urine, although the disorders are not caused by defects in the glycine cleavage system. Glycine encephalopathy is unique in the fact that levels of glycine are disproportionately elevated in CSF (in addition to blood and urine), whereas CSF glycine levels are normal or near-normal in patients with inherited organic acidurias.
Glycine metabolism
Glycine is metabolized in the body to end products of carbon dioxideCarbon dioxide
Carbon dioxide is a naturally occurring chemical compound composed of two oxygen atoms covalently bonded to a single carbon atom...
and ammonia
Ammonia
Ammonia is a compound of nitrogen and hydrogen with the formula . It is a colourless gas with a characteristic pungent odour. Ammonia contributes significantly to the nutritional needs of terrestrial organisms by serving as a precursor to food and fertilizers. Ammonia, either directly or...
. The glycine cleavage system
Glycine cleavage system
The glycine cleavage system is also known as the glycine decarboxylase complex or GCS. The system is a series of enzymes that are triggered in response to high concentrations of the amino acid glycine. The glycine cleavage system is composed of four proteins: the T-protein, P-protein, L-protein,...
, which is responsible for glycine metabolism in the mitochondrion
Mitochondrion
In cell biology, a mitochondrion is a membrane-enclosed organelle found in most eukaryotic cells. These organelles range from 0.5 to 1.0 micrometers in diameter...
is made up of four protein subunits, the P-protein, H-protein, T-protein and L-protein.
Classification
There are several different forms of glycine encephalopathy, which can be distinguished by the age of onset, as well as the types and severity of symptoms. All forms of glycine encephalopathy present with only neurological symptoms, including mental retardationMental retardation
Mental retardation is a generalized disorder appearing before adulthood, characterized by significantly impaired cognitive functioning and deficits in two or more adaptive behaviors...
, hypotonia
Hypotonia
Hypotonia is a state of low muscle tone , often involving reduced muscle strength. Hypotonia is not a specific medical disorder, but a potential manifestation of many different diseases and disorders that affect motor nerve control by the brain or muscle strength...
, seizures and brain malformations.
With the classical, or neonatal presentation of glycine encephalopathy, the infant is born after an unremarkable pregnancy, but presents with lethargy, hypotonia
Hypotonia
Hypotonia is a state of low muscle tone , often involving reduced muscle strength. Hypotonia is not a specific medical disorder, but a potential manifestation of many different diseases and disorders that affect motor nerve control by the brain or muscle strength...
, seizures and myoclonic jerks, which can progress to apnea
Apnea
Apnea, apnoea, or apnœa is a term for suspension of external breathing. During apnea there is no movement of the muscles of respiration and the volume of the lungs initially remains unchanged...
requiring artificial ventilation, and often death. Apneic patients can regain spontaneous respiration in their second to third week of life. After recovery from the initial episode, patients have intractable seizures and profound mental retardation
Mental retardation
Mental retardation is a generalized disorder appearing before adulthood, characterized by significantly impaired cognitive functioning and deficits in two or more adaptive behaviors...
, remaining developmentally delayed. Some mothers comment retrospectively that they noticed rhythmic hiccuping during pregnancy. Patients with the infantile form of glycine encephalopathy do not show lethargy and coma in the neonatal period, but often have a history of hypotonia. They often have seizures, which can range in severity and responsiveness to treatment, and are typically developmentally delayed. Glycine encephalopathy can also present as a milder form with episodic seizures, ataxia
Ataxia
Ataxia is a neurological sign and symptom that consists of gross lack of coordination of muscle movements. Ataxia is a non-specific clinical manifestation implying dysfunction of the parts of the nervous system that coordinate movement, such as the cerebellum...
, movement disorders, and gaze palsy during febrile illness. These patients are also developmentally delayed, to varying degrees. In the later onset form, patients typically have normal intellectual function, but present with spastic diplegia
Spastic diplegia
Spastic diplegia, historically known as Little's Disease, is a form of cerebral palsy that is a neuromuscular condition of hypertonia and spasticity in the muscles of the lower extremities of the human body, usually those of the legs, hips and pelvis...
and optic atrophy.
Transient neonatal hyperglycinemia has been described in a very small number of cases. Initially, these patients present with the same symptoms and laboratory results that are seen in the classical presentation. The levels of glycine in plasma and cerebrospinal fluid normalize within eight weeks, and in five of six cases there were no neurological issues at follow-up times up to thirteen years. A single patient was severely retarded at nine months. An immature glycine cleavage system in the brain and liver is suspected as the cause of transient neonatal hyperglicinemia.
Genetics
Phenylketonuria
Phenylketonuria is an autosomal recessive metabolic genetic disorder characterized by a mutation in the gene for the hepatic enzyme phenylalanine hydroxylase , rendering it nonfunctional. This enzyme is necessary to metabolize the amino acid phenylalanine to the amino acid tyrosine...
. It is caused by a defect in the glycine cleavage system
Glycine cleavage system
The glycine cleavage system is also known as the glycine decarboxylase complex or GCS. The system is a series of enzymes that are triggered in response to high concentrations of the amino acid glycine. The glycine cleavage system is composed of four proteins: the T-protein, P-protein, L-protein,...
(GCS), which is made up of four protein subunits. Each of these four subunits is encoded by a separate gene
Gene
A gene is a molecular unit of heredity of a living organism. It is a name given to some stretches of DNA and RNA that code for a type of protein or for an RNA chain that has a function in the organism. Living beings depend on genes, as they specify all proteins and functional RNA chains...
. Defects in three of these four genes have been linked to glycine encephalopathy.
| Gene | Name | Percent |
|---|---|---|
| GLDC | encodes the "glycine dehydrogenase" subunit, also called "glycine decarboxylase" | About 70-75% of cases of glycine encephalopathy result from mutations in the GLDC gene. |
| GCST or AMT Aminomethyltransferase Aminomethyltransferase is an enzyme that catabolizes the creation of methylenetetrahydrofolate. It is part of the glycine decarboxylase complex.... |
encoding the "aminomethyltransferase" subunit | About 20% of cases are caused by mutations in the AMT gene. |
| GCSH GCSH Glycine cleavage system H protein, mitochondrial is a protein that in humans is encoded by the GCSH gene.- Function :The enzyme system for cleavage of glycine , which is confined to the mitochondria, is composed of 4 protein components: P protein , H protein Glycine cleavage system H protein,... |
encoding the subunit "glycine cleavage system protein H" | Mutations in the GCSH gene account for less than 1% of cases. |
There is a fourth unit in the complex, dihydrolipoamide dehydrogenase
Dihydrolipoamide dehydrogenase
Dihydrolipoamide dehydrogenase , also known as dihydrolipoyl dehydrogenase, mitochondrial, is an enzyme that in humans is encoded by the DLD gene. DLD is a flavoprotein enzyme that degrades lipoamide, and produces dihydrolipoamide....
or GCSL. However, to date there have been no mutations in GCSL found to be associated with glycine encephalopathy.
A small percentage of affected individuals do not have a detectable mutation in any of the three genes (listed above) but still have defective glycine-cleavage enzymatic activity. It is thought that these patients may have mutations in the genes encoding one of the cofactors associated with the GCS complex.
Defects in the GSC proteins can prevent the complex from functioning properly or can prevent the GCS complex from forming entirely. When the complex is unable to break down glycine properly, this causes excess glycine to build up to toxic levels in the body's organs and tissues. Damage caused by harmful levels of glycine in the brain and cerebrospinal fluid
Cerebrospinal fluid
Cerebrospinal fluid , Liquor cerebrospinalis, is a clear, colorless, bodily fluid, that occupies the subarachnoid space and the ventricular system around and inside the brain and spinal cord...
is responsible for the characteristic seizures, breathing difficulties, movement disorders, and mental retardation.
This disorder is inherited in an autosomal recessive pattern. The term "autosomal" signifies that the gene associated with the disorder is located on an autosome
Autosome
An autosome is a chromosome that is not a sex chromosome, or allosome; that is to say, there is an equal number of copies of the chromosome in males and females. For example, in humans, there are 22 pairs of autosomes. In addition to autosomes, there are sex chromosomes, to be specific: X and Y...
. In an autosomal recessive inheritance pattern, two defective copies of the gene (one inherited from each parent) are required in order for a child to be born with the disorder. Therefore, each parent of an individual with an autosomal recessive disorder has at least one defective copy of the gene. With autosomal recessive disorders, individuals with only one copy of a defective gene (heterozygotes) are considered "carriers" for the disorder. Carriers usually do not show signs or symptoms of the disorder.