

N-Electron Valence state Perturbation Theory
Encyclopedia
In quantum chemistry
, n-electron valence state perturbation theory (NEVPT) is a perturbative treatment
applicable to multireference
CASCI-type
wavefunction
s. It can be considered as a generalization of the well-known second-order Møller-Plesset perturbation theory
to multireference Complete Active Space cases. The theory is directly integrated into the quantum chemistry package DALTON
.
The research performed into the development of this theory lead to various implementations. The theory here presented refers to the deployment for the Single-State NEVPT, where the perturbative correction is applied to a single electronic state.
Research implementations has been also developed for Quasi-Degenerate cases, where a set of electronic states undergo the perturbative correction at the same time, allowing interaction among themselves. The theory development makes use of the quasi-degenerate formalism by Lindgren and the Hamiltonian multipartitioning technique from Zaitsevskii and Malrieu.
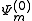 be a zero-order CASCI wavefunction, defined as a linear combination of Slater determinant
be a zero-order CASCI wavefunction, defined as a linear combination of Slater determinant
s

obtained diagonalizing the true Hamiltonian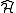 inside the CASCI space
inside the CASCI space
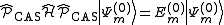
where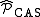 is the projector inside the CASCI space.
is the projector inside the CASCI space.
It is possible to define perturber wavefunctions in NEVPT as zero-order wavefunctions of the outer space (external to CAS) where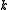 electrons are removed from the inactive part (core and virtual orbitals) and added to the valence part (active orbitals). At second order of perturbation
electrons are removed from the inactive part (core and virtual orbitals) and added to the valence part (active orbitals). At second order of perturbation 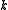 . Decomposing the zero-order CASCI wavefunction as an antisymmetrized product of the inactive part
. Decomposing the zero-order CASCI wavefunction as an antisymmetrized product of the inactive part 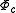 and a valence part
and a valence part 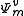
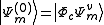
then the perturber wavefunctions can be written as
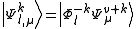
The pattern of inactive orbitals involved in the procedure can be grouped as a collective index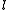 , so to represent the various perturber wavefunctions as
, so to represent the various perturber wavefunctions as 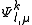 , with
, with 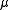 an enumerator index for the different wavefunctions. The number of these functions is relative to the degree of contraction of the resulting perturbative space.
an enumerator index for the different wavefunctions. The number of these functions is relative to the degree of contraction of the resulting perturbative space.
Supposing indexes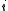 and
and 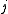 referring to core orbitals,
referring to core orbitals,  and
and 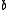 referring to active orbitals and
referring to active orbitals and  and
and  referring to virtual orbitals, the possible excitation schemes are:
referring to virtual orbitals, the possible excitation schemes are:
These cases always represent situations where interclass electronic excitations happen. Other three excitation schemes involve a single interclass excitation plus an intraclass excitation internal to the active space:
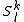 defined by those determinants with given k and l labels. It is interesting to note that the determinants characterizing these spaces can be written as a partition comprising the same inactive (core + virtual) part
defined by those determinants with given k and l labels. It is interesting to note that the determinants characterizing these spaces can be written as a partition comprising the same inactive (core + virtual) part 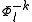 and all possible valence (active) parts
and all possible valence (active) parts 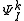
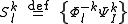
The full dimensionality of these spaces can be exploited to obtain the definition of the perturbers, by diagonalizing the Hamiltonian inside them
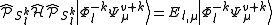
This procedure is impractical given its high computational cost: for each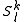 space, a diagonalization of the true Hamiltonian must be performed. Computationally, is preferable to improve the theoretical development making use of the modified Dyall's Hamiltonian
space, a diagonalization of the true Hamiltonian must be performed. Computationally, is preferable to improve the theoretical development making use of the modified Dyall's Hamiltonian 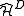 . This Hamiltonian behaves like the true Hamiltonian inside the CAS space, having the same eigenvalues and eigenvectors of the true Hamiltonian projected onto the CAS space. Also, given the decomposition for the wavefunction defined before, the action of the Dyall's Hamiltonian can be partitioned into
. This Hamiltonian behaves like the true Hamiltonian inside the CAS space, having the same eigenvalues and eigenvectors of the true Hamiltonian projected onto the CAS space. Also, given the decomposition for the wavefunction defined before, the action of the Dyall's Hamiltonian can be partitioned into
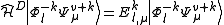
stripping out the constant contribution of the inactive part and leaving a
subsystem to be solved for the valence part
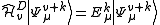
The total energy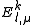 is the sum of
is the sum of 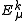 and the energies of the orbitals involved in the definition of the inactive part
and the energies of the orbitals involved in the definition of the inactive part 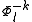 . This introduces the possibility to perform a single diagonalization of the valence Dyall's Hamiltonian on the CASCI zero-order wavefunction and evaluate the perturber energies using the property depicted above.
. This introduces the possibility to perform a single diagonalization of the valence Dyall's Hamiltonian on the CASCI zero-order wavefunction and evaluate the perturber energies using the property depicted above.
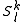 , leading to the Strongly Contracted (SC) scheme. A set of perturbative operators are used to produce a single function for each space, defined as the projection inside each space
, leading to the Strongly Contracted (SC) scheme. A set of perturbative operators are used to produce a single function for each space, defined as the projection inside each space 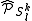 of the application of the Hamiltonian to the contracted zero order wavefunction. In other words
of the application of the Hamiltonian to the contracted zero order wavefunction. In other words
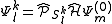
where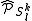 is the projector onto the subspace. This can be equivalently written as the application of a specific part of the Hamiltonian to the zero-order wavefunction
is the projector onto the subspace. This can be equivalently written as the application of a specific part of the Hamiltonian to the zero-order wavefunction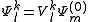
For each space, appropriate operators can be devised. We will not present their definition, as it could result overkilling. Suffice to say that the resulting perturbers are not normalized, and their norm
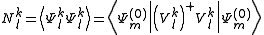
plays an important role in the Strongly Contracted development. To evaluate these norms, the spinless density matrix of rank not higher than three between the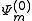 functions are needed.
functions are needed.
An important property of the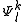 is that any other function of the space
is that any other function of the space 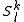 which is orthogonal to
which is orthogonal to 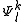 do not interact with the zero-order wavefunction through the true Hamiltonian. It is possible to use the
do not interact with the zero-order wavefunction through the true Hamiltonian. It is possible to use the 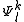 functions as a basis set for the expansion of the first-order correction to the wavefunction, and also for the expression of the zero-order Hamiltonian by means of a spectral decomposition
functions as a basis set for the expansion of the first-order correction to the wavefunction, and also for the expression of the zero-order Hamiltonian by means of a spectral decomposition
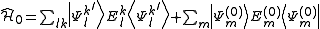
where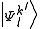 are the normalized
are the normalized 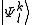 .
.
The expression for the first-order correction to the wavefunction is therefore
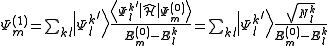
and for the energy is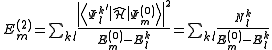
It is important to note that this result still misses a definition of the perturber energies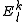 , which can be defined in a computationally advantageous approach by means of the Dyall's Hamiltonian
, which can be defined in a computationally advantageous approach by means of the Dyall's Hamiltonian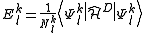
leading to
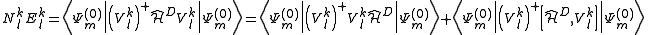
Developing the first term and extracting the inactive part of the Dyall's Hamiltonian it can be obtained
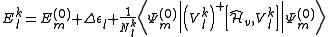
with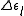 equal to the sum of the orbital energies of the newly occupied virtual orbitals minus the orbital energies of the unoccupied core orbitals.
equal to the sum of the orbital energies of the newly occupied virtual orbitals minus the orbital energies of the unoccupied core orbitals.
The term that still need to be evaluated is the braket involving the commutator. This can be obtained developing each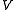 operator and substituting. To obtain the final result is necessary to evaluate Koopmans matrices and density matrices involving only active indexes. An interesting case is represented by the contribution for the
operator and substituting. To obtain the final result is necessary to evaluate Koopmans matrices and density matrices involving only active indexes. An interesting case is represented by the contribution for the 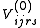 case, which is trivial and can be demonstrated identical to the Møller-Plesset second-order contribution
case, which is trivial and can be demonstrated identical to the Møller-Plesset second-order contribution
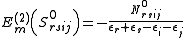
NEVPT2 can therefore be seen as a generalized form of MP2 to multireference wavefunctions.
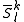 of
of  with dimensionality higher than one (like in case of the Strongly Contracted approach). To define this subspace, a set of functions
with dimensionality higher than one (like in case of the Strongly Contracted approach). To define this subspace, a set of functions 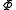 is generated by means of the
is generated by means of the 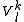 operators, after decontraction of their formulation. For example, in the case of the
operators, after decontraction of their formulation. For example, in the case of the 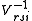 operator
operator
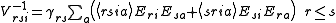
The Partially Contracted approach makes use of functions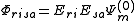 and
and 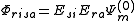 . These functions must be orthonormalized and purged of linear dependencies which may arise. The resulting set spans the
. These functions must be orthonormalized and purged of linear dependencies which may arise. The resulting set spans the 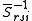 space.
space.
Once all the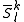 spaces have been defined, we can obtain as usual a set of perturbers from the diagonalization of the Hamiltonian (true or Dyall) inside this space
spaces have been defined, we can obtain as usual a set of perturbers from the diagonalization of the Hamiltonian (true or Dyall) inside this space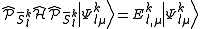
As usual, the evaluation of the Partially Contracted perturbative correction by means of the Dyall Hamiltonian involves simply manageable entities for nowadays computers.
Although the Strongly Contracted approach makes use of a perturbative space with very low flexibility, in general it provides values in very good agreement with those obtained by the more decontracted space defined for the Partially Contracted approach. This can be probably explained by the fact that the Strongly Contracted perturbers are a good average of the totally decontracted perturbative space.
It should also be noted that the Partially Contracted evaluation has a very little overhead in computational cost with respect to the Strongly Contracted one, therefore they are normally evaluated together.
Quantum chemistry
Quantum chemistry is a branch of chemistry whose primary focus is the application of quantum mechanics in physical models and experiments of chemical systems...
, n-electron valence state perturbation theory (NEVPT) is a perturbative treatment
Perturbation theory (quantum mechanics)
In quantum mechanics, perturbation theory is a set of approximation schemes directly related to mathematical perturbation for describing a complicated quantum system in terms of a simpler one. The idea is to start with a simple system for which a mathematical solution is known, and add an...
applicable to multireference
Multireference configuration interaction
In quantum chemistry, the multireference configuration interaction method consists in a configuration interaction expansion of the eigenstates of the electronic molecular Hamiltonian in a set of Slater determinants which correspond to excitations of the ground state electronic configuration but...
CASCI-type
Complete active space
In quantum chemistry, a complete active space is a type of classification of molecular orbitals. Spatial orbitals are classified as belonging to three classes:* core, always hold two electrons* active, partially occupied orbitals...
wavefunction
Wavefunction
Not to be confused with the related concept of the Wave equationA wave function or wavefunction is a probability amplitude in quantum mechanics describing the quantum state of a particle and how it behaves. Typically, its values are complex numbers and, for a single particle, it is a function of...
s. It can be considered as a generalization of the well-known second-order Møller-Plesset perturbation theory
Møller-Plesset perturbation theory
Møller–Plesset perturbation theory is one of several quantum chemistry post-Hartree–Fock ab initio methods in the field of computational chemistry...
to multireference Complete Active Space cases. The theory is directly integrated into the quantum chemistry package DALTON
DALTON
Dalton is an ab initio quantum chemistry software program. It is capable of calculating various molecular properties using the Hartree–Fock, MP2, MCSCF and coupled cluster theories. Version 2.0 of DALTON added support for density functional theory calculations...
.
The research performed into the development of this theory lead to various implementations. The theory here presented refers to the deployment for the Single-State NEVPT, where the perturbative correction is applied to a single electronic state.
Research implementations has been also developed for Quasi-Degenerate cases, where a set of electronic states undergo the perturbative correction at the same time, allowing interaction among themselves. The theory development makes use of the quasi-degenerate formalism by Lindgren and the Hamiltonian multipartitioning technique from Zaitsevskii and Malrieu.
Theory
Let be a zero-order CASCI wavefunction, defined as a linear combination of Slater determinantSlater determinant
In quantum mechanics, a Slater determinant is an expression that describes the wavefunction of a multi-fermionic system that satisfies anti-symmetry requirements and consequently the Pauli exclusion principle by changing sign upon exchange of fermions . It is named for its discoverer, John C...
s
obtained diagonalizing the true Hamiltonian
inside the CASCI spacewhere
is the projector inside the CASCI space.It is possible to define perturber wavefunctions in NEVPT as zero-order wavefunctions of the outer space (external to CAS) where
electrons are removed from the inactive part (core and virtual orbitals) and added to the valence part (active orbitals). At second order of perturbation . Decomposing the zero-order CASCI wavefunction as an antisymmetrized product of the inactive part and a valence part then the perturber wavefunctions can be written as
The pattern of inactive orbitals involved in the procedure can be grouped as a collective index
, so to represent the various perturber wavefunctions as , with an enumerator index for the different wavefunctions. The number of these functions is relative to the degree of contraction of the resulting perturbative space.Supposing indexes
and referring to core orbitals, and referring to active orbitals and and referring to virtual orbitals, the possible excitation schemes are:- two electrons from core orbitals to virtual orbitals (the active space is not enriched nor depleted of electrons, therefore
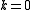 )
) - one electron from a core orbital to a virtual orbital, and one electron from a core orbital to an active orbital (the active space is enriched with one electron, therefore
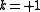 )
) - one electron from a core orbital to a virtual orbital, and one electron from an active orbital to a virtual orbital (the active space is depleted with one electron, therefore
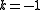 )
) - two electrons from core orbitals to active orbitals (active space enriched with two electrons,
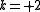 )
) - two electrons from active orbitals to virtual orbitals (active space depleted with two electrons,
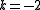 )
)
These cases always represent situations where interclass electronic excitations happen. Other three excitation schemes involve a single interclass excitation plus an intraclass excitation internal to the active space:
- one electron from a core orbital to a virtual orbital, and an internal active-active excitation (
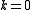 )
) - one electron from a core orbital to an active orbital, and an internal active-active excitation (
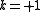 )
) - one electron from an active orbital to a virtual orbital, and an internal active-active excitation (
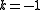 )
)
Totally Uncontracted Approach
A possible approach is to define the perturber wavefunctions into Hilbert spaces defined by those determinants with given k and l labels. It is interesting to note that the determinants characterizing these spaces can be written as a partition comprising the same inactive (core + virtual) part and all possible valence (active) parts The full dimensionality of these spaces can be exploited to obtain the definition of the perturbers, by diagonalizing the Hamiltonian inside them
This procedure is impractical given its high computational cost: for each
space, a diagonalization of the true Hamiltonian must be performed. Computationally, is preferable to improve the theoretical development making use of the modified Dyall's Hamiltonian . This Hamiltonian behaves like the true Hamiltonian inside the CAS space, having the same eigenvalues and eigenvectors of the true Hamiltonian projected onto the CAS space. Also, given the decomposition for the wavefunction defined before, the action of the Dyall's Hamiltonian can be partitioned intostripping out the constant contribution of the inactive part and leaving a
subsystem to be solved for the valence part
The total energy
is the sum of and the energies of the orbitals involved in the definition of the inactive part . This introduces the possibility to perform a single diagonalization of the valence Dyall's Hamiltonian on the CASCI zero-order wavefunction and evaluate the perturber energies using the property depicted above.Strongly Contracted Approach
A different choice in the development of the NEVPT approach is to choose a single function for each space, leading to the Strongly Contracted (SC) scheme. A set of perturbative operators are used to produce a single function for each space, defined as the projection inside each space of the application of the Hamiltonian to the contracted zero order wavefunction. In other wordswhere
is the projector onto the subspace. This can be equivalently written as the application of a specific part of the Hamiltonian to the zero-order wavefunctionFor each space, appropriate operators can be devised. We will not present their definition, as it could result overkilling. Suffice to say that the resulting perturbers are not normalized, and their norm
plays an important role in the Strongly Contracted development. To evaluate these norms, the spinless density matrix of rank not higher than three between the
functions are needed.An important property of the
is that any other function of the space which is orthogonal to do not interact with the zero-order wavefunction through the true Hamiltonian. It is possible to use the functions as a basis set for the expansion of the first-order correction to the wavefunction, and also for the expression of the zero-order Hamiltonian by means of a spectral decompositionwhere
are the normalized .The expression for the first-order correction to the wavefunction is therefore
and for the energy is
It is important to note that this result still misses a definition of the perturber energies
, which can be defined in a computationally advantageous approach by means of the Dyall's Hamiltonianleading to
Developing the first term and extracting the inactive part of the Dyall's Hamiltonian it can be obtained
with
equal to the sum of the orbital energies of the newly occupied virtual orbitals minus the orbital energies of the unoccupied core orbitals.The term that still need to be evaluated is the braket involving the commutator. This can be obtained developing each
operator and substituting. To obtain the final result is necessary to evaluate Koopmans matrices and density matrices involving only active indexes. An interesting case is represented by the contribution for the case, which is trivial and can be demonstrated identical to the Møller-Plesset second-order contributionNEVPT2 can therefore be seen as a generalized form of MP2 to multireference wavefunctions.
Partially Contracted Approach
An alternative approach, named Partially Contracted (PC) is to define the perturber wavefunctions in a subspace of with dimensionality higher than one (like in case of the Strongly Contracted approach). To define this subspace, a set of functions is generated by means of the operators, after decontraction of their formulation. For example, in the case of the operatorThe Partially Contracted approach makes use of functions
and . These functions must be orthonormalized and purged of linear dependencies which may arise. The resulting set spans the space.Once all the
spaces have been defined, we can obtain as usual a set of perturbers from the diagonalization of the Hamiltonian (true or Dyall) inside this spaceAs usual, the evaluation of the Partially Contracted perturbative correction by means of the Dyall Hamiltonian involves simply manageable entities for nowadays computers.
Although the Strongly Contracted approach makes use of a perturbative space with very low flexibility, in general it provides values in very good agreement with those obtained by the more decontracted space defined for the Partially Contracted approach. This can be probably explained by the fact that the Strongly Contracted perturbers are a good average of the totally decontracted perturbative space.
It should also be noted that the Partially Contracted evaluation has a very little overhead in computational cost with respect to the Strongly Contracted one, therefore they are normally evaluated together.
Properties
NEVPT is blessed with many important properties, making the approach very solid and reliable. These properties arise both from the theoretical approach used and on the Dyall's Hamiltonian particular structure:- Size consistencySize consistencyIn quantum chemistry, size consistency is a property that guarantees the consistency of the energy behavior when interaction between the involved molecular system is nullified ....
: NEVPT is size consistentSize consistencyIn quantum chemistry, size consistency is a property that guarantees the consistency of the energy behavior when interaction between the involved molecular system is nullified ....
(strict separable). Briefly, if A and B are two non-interacting systems, the energy of the supersystem A-B is equal to the sum of the energy of A plus the energy of B taken by themselves (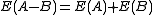 ). This property is of particular importance to obtain correctly behaving dissociation curves.
). This property is of particular importance to obtain correctly behaving dissociation curves.
- Absence of intruder stateIntruder stateIn quantum and theoretical chemistry, an intruder state is a particular situation arising in perturbative evaluations, where the energy of the perturbers is comparable in magnitude to the energy associated to the zero order wavefunction. In this case, a divergent behavior occurs, due to the nearly...
s: in perturbation theory, divergencies can occur if the energy of some perturber happens to be nearly equal to the energy of the zero-order wavefunction. This situation, which is due to the presence of an energy difference at the denominator, can be avoided if the energies associated to the perturbers are guaranteed to be never nearly equal to the zero-order energy. NEVPT satisfies this requirement.
- Invariance under active orbital rotation: The NEVPT results are stable if an intraclass active-active orbital mixing occurs. This arises both from the structure of the Dyall Hamiltonian and the properties of a CASSCF wavefunction. This property has been also extended to the intraclass core-core and virtual-virtual mixing, thanks to the Non Canonical NEVPT approach, allowing to apply a NEVPT evaluation without performing an orbital canonization (which is required, as we saw previously)
- Spin purity is guaranteed: The resulting wavefunctions are guaranteed to be spin pure, due to the spin-free formalism.
- Efficiency: although not a formal theoretical property, computational efficiency is highly important for the evaluation on medium-size molecular systems. The current limit of the NEVPT application is largely dependent on the feasibility of the previous CASSCF evaluation, which scales factorially with respect to the active space size. The NEVPT implementation using the Dyall's Hamiltonian involves the evaluation of Koopmans' matrices and density matrices up to the four-particle density matrix spanning only active orbitals. This is particularly convenient, given the small size of currently used active spaces.
- Partitioning into additive classes: The perturbative correction to the energy is additive on eight different contributions. Although the evaluation of each contribution has a different computational cost, this fact can be used to improve performance, by parallelizing each contribution to a different processor.
See also
- Electron correlation
- Perturbation theory (quantum mechanics)Perturbation theory (quantum mechanics)In quantum mechanics, perturbation theory is a set of approximation schemes directly related to mathematical perturbation for describing a complicated quantum system in terms of a simpler one. The idea is to start with a simple system for which a mathematical solution is known, and add an...
- Post-Hartree-FockPost-Hartree-FockIn computational chemistry, post-Hartree–Fock methods are the set of methods developed to improve on the Hartree–Fock , or self-consistent field method...