

Sanfilippo syndrome
Encyclopedia
Sanfilippo syndrome, or Mucopolysaccharidosis
III (MPS-III) is a rare autosomal recessive lysosomal storage disease
. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan
heparan sulfate (which is found in the extra-cellular matrix and on cell surface glycoproteins).
Although undegraded heparan sulfate is the primary stored substrate, glycolipids such as gangliosides are also stored despite no genetic defect in the enzymes associated with their breakdown.
The condition is named for Sylvester Sanfilippo
, the pediatrician who first described the disease.
The Australian study estimated the following incidences for each subtype of Sanfilippo syndrome:
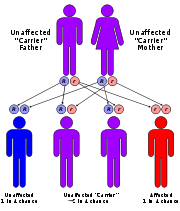 The four types of MPS-III are due to specific enzyme deficiencies affecting the breakdown of heparan sulfate, which then builds up in various organs. All four types have autosomal recessive inheritance.
The four types of MPS-III are due to specific enzyme deficiencies affecting the breakdown of heparan sulfate, which then builds up in various organs. All four types have autosomal recessive inheritance.
The disease manifests in young children. Affected infants are apparently normal, although some mild facial dysmorphism may be noticeable. The stiff joints, hirsuteness and coarse hair typical of other mucopolysaccharidoses are usually not present until late in the disease. After an initial symptom-free interval, patients usually present with a slowing of development and/or behavioral problems, followed by progressive intellectual decline resulting in severe dementia and progressive motor disease. Acquisition of speech is often slow and incomplete. The disease progresses to increasing behavioural disturbance including temper tantrums, hyperactivity, destructiveness, aggressive behaviour, pica
and sleep disturbance. As affected children have normal muscle strength and mobility, the behavioural disturbances are very difficult to manage. The disordered sleep in particular presents a significant problem to care providers. In the final phase of the illness, children become increasingly immobile and unresponsive, often require wheelchairs, and develop swallowing difficulties and seizures. The life-span of an affected child does not usually extend beyond late teens to early twenties.
Although the clinical features of the disease are mainly neurological, patients may also develop diarrhea, carious teeth, and an enlarged liver and spleen. There is a broad range of clinical severity. The disease may very rarely present later in life as a psychotic episode. [needs citation]
Of all the MPS diseases, MPS III produces the mildest physical abnormalities. It is important, however, that simple and treatable conditions such as ear infections and toothaches not be overlooked because of behavior problems that make examination difficult. Children with MPS III often have an increased tolerance of pain. Bumps and bruises or ear infections that would be painful for other children often go unnoticed in children with MPS III. Parents may need to search for a doctor with the patience and interest in treating a child with a long-term illness. Some children with MPS III may have a blood-clotting problem during and after surgery. http://mps.csupportinc.com/files/booklet_MPS_III_v6.pdf
The diagnosis may be confirmed by assay of enzyme levels in tissue samples and gene sequencing. Prenatal diagnosis is possible.
and therefore cannot treat the neurological manifestations of the disease.
Along with many other lysosomal storage diseases, MPS-III exists as a model of a monogenetic disease involving the central nervous system. Several promising therapies are in development. Gene therapy
is under investigation for MPS-III in animal models. Other potential therapies include chemical modification of deficient enzymes to allow them to penetrate the blood-brain barrier
, stabilisation of abnormal but active enzyme to prevent its degradation, and implantation of stem cell
s strongly expressing the missing enzyme. For any future treatment to be successful, it must be administered as early as possible. Currently MPS-III is mainly diagnosed clinically, by which stage it is probably too late for any treatment to be very effective. Neonatal screening programs would provide the earliest possible diagnosis.
Group of Grzegorz Węgrzyn, from Department of Molecular Biology of University of Gdansk, Poland, found that the flavonoid genistein
decreases pathological cumulation of glycosaminoglycan
s in Sanfilippo syndrome. In vitro, animal studies and clinical experiments suggest that the symptoms of the disease may be alleviated by adequate dose of genistein
. Genistein
was found to possess both health supporting but also toxic properties.
Mucopolysaccharidosis
Mucopolysaccharidoses are a group of metabolic disorders caused by the absence or malfunctioning of lysosomal enzymes needed to break down molecules called glycosaminoglycans - long chains of sugar carbohydrates in each of our cells that help build bone, cartilage, tendons, corneas, skin and...
III (MPS-III) is a rare autosomal recessive lysosomal storage disease
Lysosomal storage disease
Lysosomal storage diseases are a group of approximately 50 rare inherited metabolic disorders that result from defects in lysosomal function...
. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan
Glycosaminoglycan
Glycosaminoglycans or mucopolysaccharides are long unbranched polysaccharides consisting of a repeating disaccharide unit. The repeating unit consists of a hexose or a hexuronic acid, linked to a hexosamine .-Production:Protein cores made in the rough endoplasmic reticulum are posttranslationally...
heparan sulfate (which is found in the extra-cellular matrix and on cell surface glycoproteins).
Although undegraded heparan sulfate is the primary stored substrate, glycolipids such as gangliosides are also stored despite no genetic defect in the enzymes associated with their breakdown.
The condition is named for Sylvester Sanfilippo
Sylvester Sanfilippo
Sylvester Sanfilippo is a pediatrician from Edina, MN, who first described a mucopolysaccharide storage disease which bears his name. Sylvester Sanfilippo was born in Rochester, New York, on January 1, 1926. After graduating from the University of Rochester in 1947, he moved to Salt Lake City to...
, the pediatrician who first described the disease.
Incidence
Incidence of Sanfilippo syndrome varies geographically, with approximately 1 case per 280,000 live births in Northern Ireland, 1 per 66,000 in Australia, and 1 per 50,000 in the Netherlands.The Australian study estimated the following incidences for each subtype of Sanfilippo syndrome:
| Sanfilippo syndrome type | Approximate incidence | Percentage of cases |
|---|---|---|
| A | 1 in 100,000 | 60% |
| B | 1 in 200,000 | 30% |
| C | 1 in 1,500,000 | 5% |
| D | 1 in 1,000,000 | 6% |
Pathophysiology
| MPS-III type | enzyme | gene location |
|---|---|---|
| MPS-III A | heparan N-sulfatase | 17q25.3 |
| MPS-III B | N-acetyl-alpha-D-glucosaminidase | 17q21 |
| MPS-III C | acetyl-CoA:alpha-glucosaminide acetyltransferase | 8p11-q13 |
| MPS-III D | N-acetylglucosamine-G-sulfate sulfatase | 12q14 |
Diagnosis and Natural History
MPS-III A, B, C and D are considered to be clinically indistinguishable, although mutations in different genes are responsible for each disease. The following discussion is therefore applicable to all four conditions.The disease manifests in young children. Affected infants are apparently normal, although some mild facial dysmorphism may be noticeable. The stiff joints, hirsuteness and coarse hair typical of other mucopolysaccharidoses are usually not present until late in the disease. After an initial symptom-free interval, patients usually present with a slowing of development and/or behavioral problems, followed by progressive intellectual decline resulting in severe dementia and progressive motor disease. Acquisition of speech is often slow and incomplete. The disease progresses to increasing behavioural disturbance including temper tantrums, hyperactivity, destructiveness, aggressive behaviour, pica
Pica (disorder)
Pica is characterized by an appetite for substances largely non-nutritive . For these actions to be considered pica, they must persist for more than one month at an age where eating such objects is considered developmentally inappropriate...
and sleep disturbance. As affected children have normal muscle strength and mobility, the behavioural disturbances are very difficult to manage. The disordered sleep in particular presents a significant problem to care providers. In the final phase of the illness, children become increasingly immobile and unresponsive, often require wheelchairs, and develop swallowing difficulties and seizures. The life-span of an affected child does not usually extend beyond late teens to early twenties.
Although the clinical features of the disease are mainly neurological, patients may also develop diarrhea, carious teeth, and an enlarged liver and spleen. There is a broad range of clinical severity. The disease may very rarely present later in life as a psychotic episode. [needs citation]
Of all the MPS diseases, MPS III produces the mildest physical abnormalities. It is important, however, that simple and treatable conditions such as ear infections and toothaches not be overlooked because of behavior problems that make examination difficult. Children with MPS III often have an increased tolerance of pain. Bumps and bruises or ear infections that would be painful for other children often go unnoticed in children with MPS III. Parents may need to search for a doctor with the patience and interest in treating a child with a long-term illness. Some children with MPS III may have a blood-clotting problem during and after surgery. http://mps.csupportinc.com/files/booklet_MPS_III_v6.pdf
The diagnosis may be confirmed by assay of enzyme levels in tissue samples and gene sequencing. Prenatal diagnosis is possible.
Treatment
Treatment remains largely supportive. The behavioral disturbances of MPS-III respond poorly to medication. If an early diagnosis is made, bone marrow replacement may be beneficial. Although the missing enzyme can be manufactured and given intravenously, it cannot penetrate the blood-brain barrierBlood-brain barrier
The blood–brain barrier is a separation of circulating blood and the brain extracellular fluid in the central nervous system . It occurs along all capillaries and consists of tight junctions around the capillaries that do not exist in normal circulation. Endothelial cells restrict the diffusion...
and therefore cannot treat the neurological manifestations of the disease.
Along with many other lysosomal storage diseases, MPS-III exists as a model of a monogenetic disease involving the central nervous system. Several promising therapies are in development. Gene therapy
Gene therapy
Gene therapy is the insertion, alteration, or removal of genes within an individual's cells and biological tissues to treat disease. It is a technique for correcting defective genes that are responsible for disease development...
is under investigation for MPS-III in animal models. Other potential therapies include chemical modification of deficient enzymes to allow them to penetrate the blood-brain barrier
Blood-brain barrier
The blood–brain barrier is a separation of circulating blood and the brain extracellular fluid in the central nervous system . It occurs along all capillaries and consists of tight junctions around the capillaries that do not exist in normal circulation. Endothelial cells restrict the diffusion...
, stabilisation of abnormal but active enzyme to prevent its degradation, and implantation of stem cell
Stem cell
This article is about the cell type. For the medical therapy, see Stem Cell TreatmentsStem cells are biological cells found in all multicellular organisms, that can divide and differentiate into diverse specialized cell types and can self-renew to produce more stem cells...
s strongly expressing the missing enzyme. For any future treatment to be successful, it must be administered as early as possible. Currently MPS-III is mainly diagnosed clinically, by which stage it is probably too late for any treatment to be very effective. Neonatal screening programs would provide the earliest possible diagnosis.
Group of Grzegorz Węgrzyn, from Department of Molecular Biology of University of Gdansk, Poland, found that the flavonoid genistein
Genistein
Genistein is one of several known isoflavones. Isoflavones, such as genistein and daidzein, are found in a number of plants including lupin, fava beans, soybeans, kudzu, and psoralea being the primary food source, also in the medicinal plant, Flemingia vestita and coffee Besides functioning as...
decreases pathological cumulation of glycosaminoglycan
Glycosaminoglycan
Glycosaminoglycans or mucopolysaccharides are long unbranched polysaccharides consisting of a repeating disaccharide unit. The repeating unit consists of a hexose or a hexuronic acid, linked to a hexosamine .-Production:Protein cores made in the rough endoplasmic reticulum are posttranslationally...
s in Sanfilippo syndrome. In vitro, animal studies and clinical experiments suggest that the symptoms of the disease may be alleviated by adequate dose of genistein
Genistein
Genistein is one of several known isoflavones. Isoflavones, such as genistein and daidzein, are found in a number of plants including lupin, fava beans, soybeans, kudzu, and psoralea being the primary food source, also in the medicinal plant, Flemingia vestita and coffee Besides functioning as...
. Genistein
Genistein
Genistein is one of several known isoflavones. Isoflavones, such as genistein and daidzein, are found in a number of plants including lupin, fava beans, soybeans, kudzu, and psoralea being the primary food source, also in the medicinal plant, Flemingia vestita and coffee Besides functioning as...
was found to possess both health supporting but also toxic properties.