

VALBOND
Encyclopedia
In molecular mechanics
, VALBOND is a method for computing the angle bending energy that is based on valence bond theory
. It is based on orbital strength functions, which are maximized when the hybrid orbitals
on the atom are orthogonal. The hybridization of the bonding orbitals are obtained from empirical formulas based on Bent's rule
, which relates the preference towards p character with electronegativity.
The VALBOND functions are suitable for describing the energy of bond angle distortion not only around the equilibrium angles, but also at very large distortions. This represents an advantage over the simpler harmonic oscillator
approximation used by many force fields, and allows the VALBOND method to handle hypervalent molecule
s and transition metal complexes
. The VALBOND energy term has been combined with force fields
such as CHARMM
and UFF to provide a complete functional form that includes also bond stretching, torsions, and non-bonded interactions.
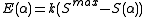 ,
,
where k is an empirical scaling factor that depends on the elements involved in the bond, Smax, the maximum strength function, is
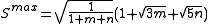
and S(α) is the strength function
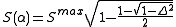
which depends on the nonorthogonality integral Δ:
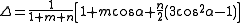
The energy contribution is added twice, once per each of the bonding orbitals involved in the angle (which may have different hybridizations and different values for k).
For non-hypervalent p-block atoms, the hybridization value n is zero (no d-orbital contribution), and m is obtained as %p(1-%p), where %p is the p character of the orbital obtained from
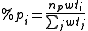
where the sum over j includes all ligands, lone pairs, and radicals on the atom, np is the "gross hybridization" (for example, for an "sp2" atom, np = 2). The weight wti depends on the two elements involved in the bond (or just one for lone pair or radicals), and represents the preference for p character of different elements. The values of the weights are empirical, but can be rationalized in terms of Bent's rule.
s (3c4e) in different ways. For example, ClF3 is represented as having one "normal" two-center bond and one 3c4e bond. There are three different configurations for ClF3, each one using a different Cl-F bond as the two-center bond. For more complicated systems the number of combinations increases rapidly; SF6 has 45 configurations.
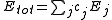
where the sum is over all configurations j, and the coefficient cj is defined by the function
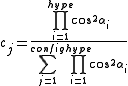
where "hype" refers to the 3c4e bonds. This function ensures that the configurations where the 3c4e bonds are linear are favored.
The energy terms are modified by multiplying them by a bond order factor, BOF, which is the product of the formal bond orders of the two bonds involved in the angle (for 3c4e bonds, the bond order is 0.5). For 3c4e bonds, the energy is calculated as
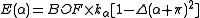
where Δ is again the non-orthogonality function, but here the angle α is offset by 180 degrees (π radians).
Finally, to ensure that the axial vs equatorial preference of different ligands in hypervalent compounds is reproduced, an "offset energy" term is subtracted. It has the form
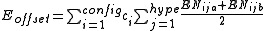
where the EN terms depend on the electronegativity
difference between the ligand and the central atom as follows:
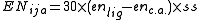
where ss is 1 if the electronegativity difference is positive and 2 if it is negative.
For p-block hypervalent molecules, d orbitals are not used, so n = 0. The p contribution m is estimated from ab initio quantum chemistry methods
and a natural bond orbital (NBO) analysis.
Molecular mechanics
Molecular mechanics uses Newtonian mechanics to model molecular systems. The potential energy of all systems in molecular mechanics is calculated using force fields...
, VALBOND is a method for computing the angle bending energy that is based on valence bond theory
Valence bond theory
In chemistry, valence bond theory is one of two basic theories, along with molecular orbital theory, that were developed to use the methods of quantum mechanics to explain chemical bonding. It focuses on how the atomic orbitals of the dissociated atoms combine to give individual chemical bonds...
. It is based on orbital strength functions, which are maximized when the hybrid orbitals
Orbital hybridisation
In chemistry, hybridisation is the concept of mixing atomic orbitals to form new hybrid orbitals suitable for the qualitative description of atomic bonding properties. Hybridised orbitals are very useful in the explanation of the shape of molecular orbitals for molecules. It is an integral part...
on the atom are orthogonal. The hybridization of the bonding orbitals are obtained from empirical formulas based on Bent's rule
Bent's rule
Bent’s rule was formulated in 1961 by American chemist, Henry Bent, to explain deviations in structures predicted from the VSEPR theory. The rule states: “atomic s character tends to concentrate in orbitals that are directed toward electropositive groups and atomic p character tends to concentrate...
, which relates the preference towards p character with electronegativity.
The VALBOND functions are suitable for describing the energy of bond angle distortion not only around the equilibrium angles, but also at very large distortions. This represents an advantage over the simpler harmonic oscillator
Harmonic oscillator
In classical mechanics, a harmonic oscillator is a system that, when displaced from its equilibrium position, experiences a restoring force, F, proportional to the displacement, x: \vec F = -k \vec x \, where k is a positive constant....
approximation used by many force fields, and allows the VALBOND method to handle hypervalent molecule
Hypervalent molecule
A hypervalent molecule is a molecule that contains one or more main group elements formally bearing more than eight electrons in their valence shells...
s and transition metal complexes
Complex (chemistry)
In chemistry, a coordination complex or metal complex, is an atom or ion , bonded to a surrounding array of molecules or anions, that are in turn known as ligands or complexing agents...
. The VALBOND energy term has been combined with force fields
Force field (chemistry)
In the context of molecular modeling, a force field refers to the form and parameters of mathematical functions used to describe the potential energy of a system of particles . Force field functions and parameter sets are derived from both experimental work and high-level quantum mechanical...
such as CHARMM
CHARMM
CHARMM is the name of a widely used set of force fields for molecular dynamics as well as the name for the molecular dynamics simulation and analysis package associated with them...
and UFF to provide a complete functional form that includes also bond stretching, torsions, and non-bonded interactions.
Non-hypervalent molecules
For an angle α between normal (non-hypervalent) bonds involving an spmdn hybrid orbital, the energy contribution is,where k is an empirical scaling factor that depends on the elements involved in the bond, Smax, the maximum strength function, is
and S(α) is the strength function
which depends on the nonorthogonality integral Δ:
The energy contribution is added twice, once per each of the bonding orbitals involved in the angle (which may have different hybridizations and different values for k).
For non-hypervalent p-block atoms, the hybridization value n is zero (no d-orbital contribution), and m is obtained as %p(1-%p), where %p is the p character of the orbital obtained from
where the sum over j includes all ligands, lone pairs, and radicals on the atom, np is the "gross hybridization" (for example, for an "sp2" atom, np = 2). The weight wti depends on the two elements involved in the bond (or just one for lone pair or radicals), and represents the preference for p character of different elements. The values of the weights are empirical, but can be rationalized in terms of Bent's rule.
Hypervalent molecules
For hypervalent molecules, the energy is represented as a combination of VALBOND configurations, which are akin to resonance structures that place three-center four-electron bondThree-center four-electron bond
The 3-center 4-electron bond is a model used to explain bonding in hypervalent molecules such as phosphorus pentafluoride, sulfur hexafluoride, the xenon fluorides, and the bifluoride ion. It is also known as the Pimentel-Rundle three-center model after the work published by George C. Pimentel in...
s (3c4e) in different ways. For example, ClF3 is represented as having one "normal" two-center bond and one 3c4e bond. There are three different configurations for ClF3, each one using a different Cl-F bond as the two-center bond. For more complicated systems the number of combinations increases rapidly; SF6 has 45 configurations.
where the sum is over all configurations j, and the coefficient cj is defined by the function
where "hype" refers to the 3c4e bonds. This function ensures that the configurations where the 3c4e bonds are linear are favored.
The energy terms are modified by multiplying them by a bond order factor, BOF, which is the product of the formal bond orders of the two bonds involved in the angle (for 3c4e bonds, the bond order is 0.5). For 3c4e bonds, the energy is calculated as
where Δ is again the non-orthogonality function, but here the angle α is offset by 180 degrees (π radians).
Finally, to ensure that the axial vs equatorial preference of different ligands in hypervalent compounds is reproduced, an "offset energy" term is subtracted. It has the form
where the EN terms depend on the electronegativity
Electronegativity
Electronegativity, symbol χ , is a chemical property that describes the tendency of an atom or a functional group to attract electrons towards itself. An atom's electronegativity is affected by both its atomic number and the distance that its valence electrons reside from the charged nucleus...
difference between the ligand and the central atom as follows:
where ss is 1 if the electronegativity difference is positive and 2 if it is negative.
For p-block hypervalent molecules, d orbitals are not used, so n = 0. The p contribution m is estimated from ab initio quantum chemistry methods
Ab initio quantum chemistry methods
Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initiowas first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited states of benzene.The background is described by Parr...
and a natural bond orbital (NBO) analysis.