

Wolman disease
Encyclopedia
Wolman DiseaseWolman Disease (also known as Wolman’s Disease, early onset LAL Deficiency, and Lysosomal acid lipase deficiency
is a rare genetic disorder caused by a deficiency of an enzyme
known as lysosomal acid lipase (LAL or LIPA). This enzyme is necessary to break down certain lipids inside the cells. Deficiency of the LAL/LIPA enzyme causes a build-up of fat in the liver, gut and other parts of the body.
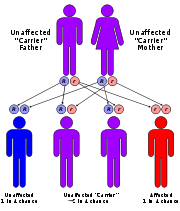 What causes Wolman Disease? Every person has two copies of the LAL gene
What causes Wolman Disease? Every person has two copies of the LAL gene
(sometimes known as the LIPA gene). One copy is inherited from the father and one from the mother. LAL Deficiency occurs when a person has defects (mutations) in both copies of the LAL gene. Each parent of a patient with LAL deficiency carries one copy of the defective LAL gene. With every pregnancy, parents with a son or daughter affected by LAL Deficiency have a 1 in 4 (25%) chance of having another affected child. A person born with defects in both LAL genes is not able to produce adequate amounts of the LAL enzyme.
Lysosomes function as recycling centers within cells breaking down unwanted materials into substances that the cells can reuse. The LAL enzyme is responsible for the breakdown of specific fats within the lysosomes. When the lysosome does not have enough LAL enzyme, it is unable to breakdown these fats.
How common is Wolman Disease? Like many rare diseases, Wolman disease may go undiagnosed or misdiagnosed which means that there is not much information in the medical literature about how common this condition is. Wolman disease affects males and females in equal numbers. In one report, 8 cases of Wolman disease were identified out more than 4.2 million births over the 16-year period. This translates to an incidence around 1 case per 500,000 live births. For a country the size of the United States, this means there may be 8 babies with Wolman disease born each year.
Are there any specific populations that get Wolman disease more frequently than others? Because Wolman disease is autosomal recessive, it is expected to occur at higher frequencies in certain populations where marriages between genetically related individuals occur (consanguinity). A higher frequency of an abnormal LAL gene has been recently described in the Iranian-Jewish population.
A diagnosis of Wolman disease may be suspected in newborn infants based upon identification of characteristic symptoms such as abnormally enlarged liver and gastrointestinal problems.
The accumulation of fat in the walls of the gut in Wolman disease leads to serious digestive problems including malabsorption
, a condition in which the gut fails to absorb nutrients and calories from food. The malabsorption associated with Wolman disease is often accompanied by persistent and often forceful vomiting, frequent diarrhea, foul-smelling and fatty stools (steatorrhea). Because of these digestive complications, affected infants usually fail to grow and gain weight at the expected rate for their age (failure to thrive).
As the disease progresses, increasing fat accumulation in the liver leads to other complications including yellowing of the skin and whites of the eyes (jaundice
), and a persistent low-grade fever.
A distinct finding associated with Wolman disease is the accumulation of chalky material (calcification
) in the adrenal gland
.
The complications of Wolman disease progress over time, eventually leading to life-threatening problems such as extremely low levels of circulating red blood cells (severe anemia
), liver dysfunction or failure, and physical wasting (cachexia
). Very few infants with Wolman disease survive beyond the first year of life.
Treatment is mainly focused on reducing specific complications and these complications may differ between patients. Treatment is often provided in specialized centers and may require input from a team of different specialists. Genetic counseling may be of benefit for affected individuals and their families. Specific interventions may include:
Investigational Therapies
Some children with Wolman Disease have had an experimental therapy called hematopoietic stem cell transplantation (HSCT), also known as bone marrow transplant
, to try to prevent the disease from getting worse.
Researchers are currently studying enzyme replacement therapy
for LSDs such as Wolman disease. Enzyme replacement therapy involves replacing a missing enzyme in individuals who are deficient or lack the particular enzyme in question. Synthetic versions of missing enzymes have been developed and used to successfully treat individuals with other LSDs including Fabry disease and Gaucher disease.
Gene therapy is also being studied as another possible approach to therapy for some LSDs. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the production of active enzyme and prevent the development and progression of the disease in question. Given the permanent transfer of the normal gene, which is able to produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a "cure." However, at this time, there are many technical difficulties to resolve before gene therapy can succeed.
Synageva BioPharma Corp. of Lexington, Massachusetts, is recruiting patients to participate in a clinical trial that evaluates an enzyme replacement therapy of Lysosomal Acid Lipase (LAL) Deficiency.
Lysosomal Acid Lipase Deficiency
Lysosomal Acid Lipase Deficiency happens when the body does not produce enough active LAL enzyme. Under normal conditions, the body produces an enzyme called lysosomal acid lipase . This enzyme plays an important role in breaking down fatty material in the body...
is a rare genetic disorder caused by a deficiency of an enzyme
Enzyme
Enzymes are proteins that catalyze chemical reactions. In enzymatic reactions, the molecules at the beginning of the process, called substrates, are converted into different molecules, called products. Almost all chemical reactions in a biological cell need enzymes in order to occur at rates...
known as lysosomal acid lipase (LAL or LIPA). This enzyme is necessary to break down certain lipids inside the cells. Deficiency of the LAL/LIPA enzyme causes a build-up of fat in the liver, gut and other parts of the body.
- Wolman disease belongs to a group of diseases known as Lysosomal Storage Disorders (LSDs).
- Lysosomes function as recycling centers within cells breaking down a number of unwanted materials into substances that the cell can reuse.
- Enzymes are highly specialized proteins within lysosomes that break down or digest particular nutrients, such as certain fats and carbohydrates.
- When these enzymes are defective or missing altogether because of genetic mutations, LSDs develop as a result of abnormal build-up of material in the body's cells.
- Wolman disease is the early onset form of LAL Deficiency.
- This form of the disease typically develops during the first few weeks or month of life.
- Late onset form which is known as Cholesteryl ester storage diseaseCholesteryl ester storage diseaseCholesteryl Ester Storage Disease is the late onset phenotype for Lysosomal Acid Lipase Deficiency, a Lysosomal storage disease, which also has an early onset phenotype known as Wolman disease that primarily affects infants. CESD can present in childhood but often goes unrecognized until...
(CESD) typically presents later in childhood or even adulthood.
Inheritance and Diagnosis
Gene
A gene is a molecular unit of heredity of a living organism. It is a name given to some stretches of DNA and RNA that code for a type of protein or for an RNA chain that has a function in the organism. Living beings depend on genes, as they specify all proteins and functional RNA chains...
(sometimes known as the LIPA gene). One copy is inherited from the father and one from the mother. LAL Deficiency occurs when a person has defects (mutations) in both copies of the LAL gene. Each parent of a patient with LAL deficiency carries one copy of the defective LAL gene. With every pregnancy, parents with a son or daughter affected by LAL Deficiency have a 1 in 4 (25%) chance of having another affected child. A person born with defects in both LAL genes is not able to produce adequate amounts of the LAL enzyme.
Lysosomes function as recycling centers within cells breaking down unwanted materials into substances that the cells can reuse. The LAL enzyme is responsible for the breakdown of specific fats within the lysosomes. When the lysosome does not have enough LAL enzyme, it is unable to breakdown these fats.
How common is Wolman Disease? Like many rare diseases, Wolman disease may go undiagnosed or misdiagnosed which means that there is not much information in the medical literature about how common this condition is. Wolman disease affects males and females in equal numbers. In one report, 8 cases of Wolman disease were identified out more than 4.2 million births over the 16-year period. This translates to an incidence around 1 case per 500,000 live births. For a country the size of the United States, this means there may be 8 babies with Wolman disease born each year.
Are there any specific populations that get Wolman disease more frequently than others? Because Wolman disease is autosomal recessive, it is expected to occur at higher frequencies in certain populations where marriages between genetically related individuals occur (consanguinity). A higher frequency of an abnormal LAL gene has been recently described in the Iranian-Jewish population.
A diagnosis of Wolman disease may be suspected in newborn infants based upon identification of characteristic symptoms such as abnormally enlarged liver and gastrointestinal problems.
- A diagnosis may be confirmed by a thorough clinical evaluation, a detailed patient history (including family history) and specialized tests that reveal absence or deficient activity of the enzyme lysosomal lipase acid (LIPA) in certain cells and tissues of the body.
- Diagnosis may also be made by confirming that there are mutations (defects) in both copies of the LIPA gene.
- Diagnosis before birth (prenatally) is possible through chorionic villus samplingChorionic villus samplingChorionic villus sampling , sometimes misspelled "chorionic villous sampling", is a form of prenatal diagnosis to determine chromosomal or genetic disorders in the fetus. It entails sampling of the chorionic villus and testing it...
(CVS) or amniocentesisAmniocentesisAmniocentesis is a medical procedure used in prenatal diagnosis of chromosomal abnormalities and fetal infections, in which a small amount of amniotic fluid, which contains fetal tissues, is sampled from the amnion or amniotic sac surrounding a developing fetus, and the fetal DNA is examined for...
.- During CVS, fetal tissue samples are removed and enzyme tests (assays) are performed on cultured tissue cells (fibroblasts) and/or white blood cells (leukocytes).
- During amniocentesisAmniocentesisAmniocentesis is a medical procedure used in prenatal diagnosis of chromosomal abnormalities and fetal infections, in which a small amount of amniotic fluid, which contains fetal tissues, is sampled from the amnion or amniotic sac surrounding a developing fetus, and the fetal DNA is examined for...
, a sample of fluid that surrounds the developing fetus is removed and studied.
Symptoms
The signs and symptoms of Wolman disease usually appear shortly after birth, typically in the first few weeks of life. Affected infants may have the following:- Feeding difficulties with frequent vomiting
- Diarrhea (loose frequent stools)
- Swelling of the abdomen (abdominal distention)
- Enlargement of the liver (hepatosplenomegalyHepatosplenomegalyHepatosplenomegaly is the simultaneous enlargement of both the liver and the spleen . Hepatosplenomegaly can occur as the result of acute viral hepatitis or infectious mononucleosis, or it can be the sign of a serious and life threatening lysosomal storage disease...
) and spleen (splenomegalySplenomegalySplenomegaly is an enlargement of the spleen. The spleen usually lies in the left upper quadrant of the human abdomen. It is one of the four cardinal signs of hypersplenism, some reduction in the number of circulating blood cells affecting granulocytes, erythrocytes or platelets in any...
) - Failure to gain weight or sometimes weight loss
The accumulation of fat in the walls of the gut in Wolman disease leads to serious digestive problems including malabsorption
Malabsorption
Malabsorption is a state arising from abnormality in absorption of food nutrients across the gastrointestinal tract.Impairment can be of single or multiple nutrients depending on the abnormality...
, a condition in which the gut fails to absorb nutrients and calories from food. The malabsorption associated with Wolman disease is often accompanied by persistent and often forceful vomiting, frequent diarrhea, foul-smelling and fatty stools (steatorrhea). Because of these digestive complications, affected infants usually fail to grow and gain weight at the expected rate for their age (failure to thrive).
As the disease progresses, increasing fat accumulation in the liver leads to other complications including yellowing of the skin and whites of the eyes (jaundice
Jaundice
Jaundice is a yellowish pigmentation of the skin, the conjunctival membranes over the sclerae , and other mucous membranes caused by hyperbilirubinemia . This hyperbilirubinemia subsequently causes increased levels of bilirubin in the extracellular fluid...
), and a persistent low-grade fever.
A distinct finding associated with Wolman disease is the accumulation of chalky material (calcification
Calcification
Calcification is the process in which calcium salts build up in soft tissue, causing it to harden. Calcifications may be classified on whether there is mineral balance or not, and the location of the calcification.-Causes:...
) in the adrenal gland
Adrenal gland
In mammals, the adrenal glands are endocrine glands that sit atop the kidneys; in humans, the right suprarenal gland is triangular shaped, while the left suprarenal gland is semilunar shaped...
.
- The adrenal glands are located on top of the kidneys and produce hormones (chemicals that help regulate various functions in the body).
- CalcificationCalcificationCalcification is the process in which calcium salts build up in soft tissue, causing it to harden. Calcifications may be classified on whether there is mineral balance or not, and the location of the calcification.-Causes:...
of the adrenal glands is not detectable by physical examination, but can be seen with an X-ray, CT scan or other imaging investigations. This finding is very useful in helping make a diagnosis as there are very few other conditions that cause vomiting, failure to gain weight and adrenal calcification. Not all cases of Wolman disease show this finding and diagnosis in these cases is more difficult and often delayed. Calcification may prevent the adrenal glands from producing enough essential hormones and can affect metabolism, blood pressure, the immune system and other vital processes of the body. It has not been definitely established in Wolman disease that the adrenal calcification causes abnormalities in adrenal function.
The complications of Wolman disease progress over time, eventually leading to life-threatening problems such as extremely low levels of circulating red blood cells (severe anemia
Anemia
Anemia is a decrease in number of red blood cells or less than the normal quantity of hemoglobin in the blood. However, it can include decreased oxygen-binding ability of each hemoglobin molecule due to deformity or lack in numerical development as in some other types of hemoglobin...
), liver dysfunction or failure, and physical wasting (cachexia
Cachexia
Cachexia or wasting syndrome is loss of weight, muscle atrophy, fatigue, weakness, and significant loss of appetite in someone who is not actively trying to lose weight...
). Very few infants with Wolman disease survive beyond the first year of life.
Prognosis
There are currently no approved treatments for Wolman disease and no treatments have been shown in clinical trials to stop or reverse the abnormalities in patients with Wolman disease.Treatment is mainly focused on reducing specific complications and these complications may differ between patients. Treatment is often provided in specialized centers and may require input from a team of different specialists. Genetic counseling may be of benefit for affected individuals and their families. Specific interventions may include:
- A change from breast or normal bottle formula to a specialized low fat formula
- Introduction of intravenous feeding (parenteral nutrition)
- Antibiotics for infections
- Steroid replacement therapy because of concerns about adrenal function
Investigational Therapies
Some children with Wolman Disease have had an experimental therapy called hematopoietic stem cell transplantation (HSCT), also known as bone marrow transplant
Bone marrow transplant
Hematopoietic stem cell transplantation is the transplantation of multipotent hematopoietic stem cell or blood, usually derived from bone marrow, peripheral blood stem cells, or umbilical cord blood...
, to try to prevent the disease from getting worse.
- Hematopoietic stem cells are specialized cells found in the bone marrow (the soft spongy material found in long bones). These blood stem cells grow and eventually develop into one of the three main types of blood cells - red blood cells, white blood cells or platelets.
- Stem cell replacement, which requires the child to be hospitalized and treated with very strong medicines before and after the procedure, has its own significant risks and potential benefits
- The healthy cells produced by the new marrow contain sufficient levels of lysosomal acid lipase required to breakdown cholesterol and triglycerides.
- Individuals with Wolman disease treated with hematopoietic stem cell transplantation have shown improvement of existing symptoms and avoidance of additional complications such as liver failure.
- HSCT for Wolmans disease at present is associated with a high risk of serious complications including death, graft-versus-host disease and other long-term and late effects.
- More research is necessary to determine the long-term safety and effectiveness of this potential therapy for infants with Wolman disease.
Researchers are currently studying enzyme replacement therapy
Enzyme replacement therapy
Enzyme replacement therapy is a medical treatment replacing an enzyme in patients in whom that particular enzyme is deficient or absent. Usually this is done by giving the patient an intravenous infusion containing the enzyme...
for LSDs such as Wolman disease. Enzyme replacement therapy involves replacing a missing enzyme in individuals who are deficient or lack the particular enzyme in question. Synthetic versions of missing enzymes have been developed and used to successfully treat individuals with other LSDs including Fabry disease and Gaucher disease.
Gene therapy is also being studied as another possible approach to therapy for some LSDs. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the production of active enzyme and prevent the development and progression of the disease in question. Given the permanent transfer of the normal gene, which is able to produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a "cure." However, at this time, there are many technical difficulties to resolve before gene therapy can succeed.
Synageva BioPharma Corp. of Lexington, Massachusetts, is recruiting patients to participate in a clinical trial that evaluates an enzyme replacement therapy of Lysosomal Acid Lipase (LAL) Deficiency.
External links
- LSD clinical trials
- Synageva BioPharma Corp.
- Clinical Trials.gov
- National Organization for Rare Disorders (NORD)
- LAL Solace (Support Organization for LAL Deficiency - Advocacy, Care and Expertise)
- Hide & Seek Foundation For Lysosomal Disease Research
- Learning Radiology.com - More information about adrenal calcification
- Radswiki.net
- Wrong Diagnosis.com
- Medlink.com