

BLOSUM
Encyclopedia
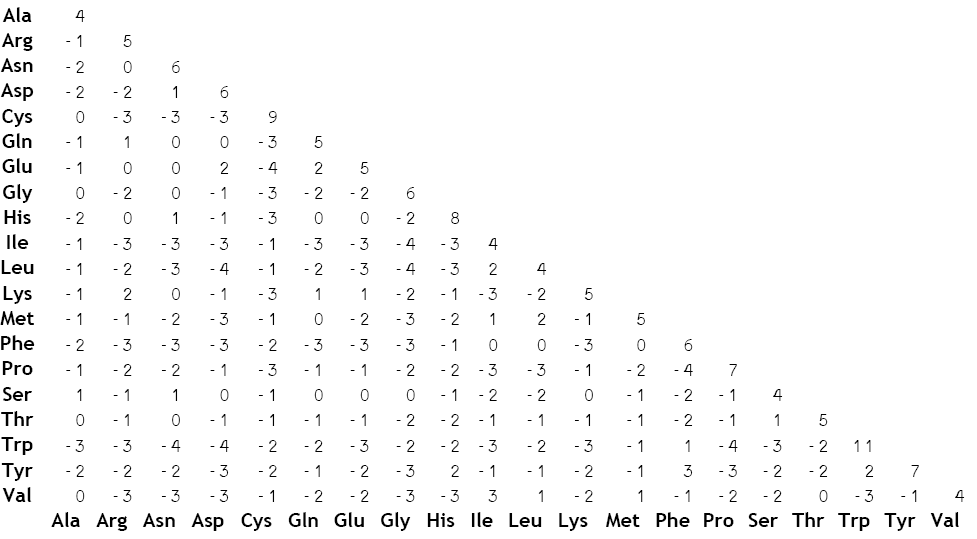
Substitution matrix
In bioinformatics and evolutionary biology, a substitution matrix describes the rate at which one character in a sequence changes to other character states over time...
used for sequence alignment
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
of protein
Protein
Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form, facilitating a biological function. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of...
s. BLOSUM matrices are used to score alignments between evolutionarily divergent protein sequences. They are based on local alignments. BLOSUM matrices were first introduced in a paper by Henikoff and Henikoff. They scanned the BLOCKS database for very conserved regions of protein families (that do not have gaps in the sequence alignment) and then counted the relative frequencies of amino acids and their substitution probabilities. Then, they calculated a log-odds
Odds ratio
The odds ratio is a measure of effect size, describing the strength of association or non-independence between two binary data values. It is used as a descriptive statistic, and plays an important role in logistic regression...
score for each of the 210 possible substitutions of the 20 standard amino acids. All BLOSUM matrices are based on observed alignments; they are not extrapolated from comparisons of closely related proteins like the PAM Matrices
Point accepted mutation
Point accepted mutation , is a set of matrices used to score sequence alignments. The PAM matrices were introduced by Margaret Dayhoff in 1978 based on 1572 observed mutations in 71 families of closely related proteins...
.
Several sets of BLOSUM matrices exist using different alignment databases, named with numbers. BLOSUM matrices with high numbers are designed for comparing closely related sequences, while those with low numbers are designed for comparing distant related sequences. For example, BLOSUM80 is used for less divergent alignments, and BLOSUM45 is used for more divergent alignments. The matrices were created by merging (clustering) all sequences that were more similar than a given percentage into one single sequence and then comparing those sequences (that were all more divergent than the given percentage value) only; thus reducing the contribution of closely related sequences. The percentage used was appended to the name, giving BLOSUM80 for example where sequences that were more than 80% identical were clustered.
Scores within a BLOSUM are log-odds scores that measure, in an alignment, the logarithm for the ratio of the likelihood of two amino acids appearing with a biological sense and the likelihood of the same amino acids appearing by chance. The matrices are based on the minimum percentage identity of the aligned protein sequence used in calculating them. Every possible identity or substitution is assigned a score based on its observed frequences in the alignment of related proteins. A positive score is given to the more likely substitutions while a negative score is given to the less likely substitutions.
To calculate a BLOSUM matrix, the following equation is used:
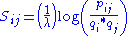
Here,
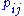 is the probability of two amino acids
is the probability of two amino acids 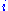 and
and 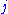 replacing each other in a homologous sequence, and
replacing each other in a homologous sequence, and 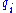 and
and 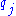 are the background probabilities of finding the amino acids
are the background probabilities of finding the amino acids 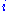 and
and 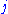 in any protein sequence at random. The factor
in any protein sequence at random. The factor 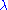 is a scaling factor, set such that the matrix contains easily computable integer values.
is a scaling factor, set such that the matrix contains easily computable integer values.An article in Nature Biotechnology revealed that the BLOSUM62 used for so many years as a standard is not exactly accurate according to the algorithm described by Henikoff and Henikoff. Surprisingly, the miscalculated BLOSUM62 improves search performance.
External links
- Page on BLOSUM
- BLOCKS WWW server
- Scoring systems for BLAST at NCBI
- [ftp://ftp.ncbi.nih.gov/blast/matrices/ Data files of BLOSUM on the NCBI FTP server].