

Substitution matrix
Encyclopedia
In bioinformatics
and evolutionary biology, a substitution matrix describes the rate at which one character in a sequence changes to other character states over time. Substitution matrices are usually seen in the context of amino acid
or DNA
sequence alignment
s, where the similarity between sequences depends on their divergence time and the substitution rates as represented in the matrix.
, from one generation to the next the amino acid sequences of an organism's proteins are gradually altered through the action of DNA mutations. For example, the sequence
ALEIRYLRD
could mutate into the sequence
ALEINYLRD
in one step, and possibly
AQEINYQRD
over a longer period of evolutionary time. Each amino acid is more or less likely to mutate into various other amino acids. For instance, a hydrophilic residue such as arginine
is more likely to be replaced another hydrophilic residue such as glutamine
, than it is to be mutated into a hydrophobic residue such as leucine
. This is primarily due to redundancy in the genetic code
, which translates similar codons into similar amino acids. Furthermore, mutating an amino acid to a residue with significantly different properties could affect the folding
and/or activity of the protein. There is therefore usually strong selective pressure to remove such mutations quickly from a population.
If we have two amino acid sequences in front of us, we should be able to say something about how likely they are to be derived from a common ancestor, or homologous
. If we can line up the two sequences using a sequence alignment
algorithm such that the mutations required to transform a hypothetical ancestor sequence into both of the current sequences would be evolutionarily plausible, then we'd like to assign a high score to the comparison of the sequences.
To this end, we will construct a 20x20 matrix where the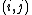 th entry is equal to the probability of the
th entry is equal to the probability of the 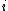 th amino acid being transformed into the
th amino acid being transformed into the 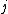 th amino acid in a certain amount of evolutionary time. There are many different ways to construct such a matrix, called a substitution matrix. Here are the most commonly used ones:
th amino acid in a certain amount of evolutionary time. There are many different ways to construct such a matrix, called a substitution matrix. Here are the most commonly used ones:
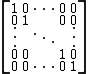
This identity matrix
will succeed in the alignment of very similar amino acid sequences but will be miserable at aligning two distantly related sequences. We need to figure out all the probabilities in a more rigorous fashion. It turns out that an empirical examination of previously aligned sequences works best.
of transformation in what are called log-odds score
s. The scores matrix S is defined as
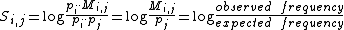
where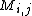 is the probability that amino acid
is the probability that amino acid 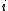 transforms into amino acid
transforms into amino acid 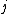 and
and 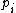 is the frequency of amino acid i. The base of the logarithm is not important, and you will often see the same substitution matrix expressed in different bases.
is the frequency of amino acid i. The base of the logarithm is not important, and you will often see the same substitution matrix expressed in different bases.
) matrix was developed by Margaret Dayhoff
in the 1970s. This matrix is calculated by observing the differences in closely related proteins. The PAM1 matrix estimates what rate of substitution would be expected if 1% of the amino acids had changed. The PAM1 matrix is used as the basis for calculating other matrices by assuming that repeated mutations would follow the same pattern as those in the PAM1 matrix, and multiple substitutions can occur at the same site. Using this logic, Dayhoff derived matrices as high as PAM250. Usually the PAM 30 and the PAM70 are used.
A matrix for divergent sequences can be calculated from a matrix for closely related sequences by taking the second matrix to a power. For instance, we can roughly approximate the WIKI2 matrix from the WIKI1 matrix by saying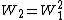 where
where 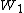 is WIKI1 and
is WIKI1 and 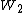 is WIKI2. This is how the PAM250 matrix is calculated.
is WIKI2. This is how the PAM250 matrix is calculated.
(BLOck SUbstitution Matrix) series of matrices rectifies this problem. Henikoff and Henikoff constructed these matrices using multiple alignments of evolutionarily divergent proteins. The probabilities used in the matrix calculation are computed by looking at "blocks" of conserved sequences found in multiple protein alignments. These conserved sequences are assumed to be of functional importance within related proteins. To reduce bias from closely related sequences, segments in a block with a sequence identity above a certain threshold were clustered giving weight to each such cluster (Henikoff and Henikoff). For the BLOSUM62 matrix, this threshold was set at 62%. Pairs frequencies were then counted between clusters, hence pairs were only counted between segments less than 62% identical. One would use a higher numbered BLOSUM matrix for aligning two closely related sequences and a lower number for more divergent sequences.
It turns out that the BLOSUM62 matrix does an excellent job detecting similarities in distant sequences, and this is the matrix used by default in most recent alignment applications such as BLAST
.
program has been demonstrated to achieve a twofold sensitivity improvement for remotely related sequences over BLAST at similar speeds (CS-BLAST
).
" is also used to indicate those substitutions that are between the two-ring purine
s (A → G and G → A) or are between the one-ring pyrimidine
s (C → T and T → C). Because these substitutions do not require a change in the number of rings, they occur more frequently than the other substitutions. "Transversion
" is the term used to indicate the slower-rate substitutions that change a purine to a pyrimidine or vice versa (A ↔ C, A ↔ T, G ↔ C, and G ↔ T).
Bioinformatics
Bioinformatics is the application of computer science and information technology to the field of biology and medicine. Bioinformatics deals with algorithms, databases and information systems, web technologies, artificial intelligence and soft computing, information and computation theory, software...
and evolutionary biology, a substitution matrix describes the rate at which one character in a sequence changes to other character states over time. Substitution matrices are usually seen in the context of amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
or DNA
DNA
Deoxyribonucleic acid is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms . The DNA segments that carry this genetic information are called genes, but other DNA sequences have structural purposes, or are involved in...
sequence alignment
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
s, where the similarity between sequences depends on their divergence time and the substitution rates as represented in the matrix.
Background
In the process of evolutionEvolution
Evolution is any change across successive generations in the heritable characteristics of biological populations. Evolutionary processes give rise to diversity at every level of biological organisation, including species, individual organisms and molecules such as DNA and proteins.Life on Earth...
, from one generation to the next the amino acid sequences of an organism's proteins are gradually altered through the action of DNA mutations. For example, the sequence
ALEIRYLRD
could mutate into the sequence
ALEINYLRD
in one step, and possibly
AQEINYQRD
over a longer period of evolutionary time. Each amino acid is more or less likely to mutate into various other amino acids. For instance, a hydrophilic residue such as arginine
Arginine
Arginine is an α-amino acid. The L-form is one of the 20 most common natural amino acids. At the level of molecular genetics, in the structure of the messenger ribonucleic acid mRNA, CGU, CGC, CGA, CGG, AGA, and AGG, are the triplets of nucleotide bases or codons that codify for arginine during...
is more likely to be replaced another hydrophilic residue such as glutamine
Glutamine
Glutamine is one of the 20 amino acids encoded by the standard genetic code. It is not recognized as an essential amino acid but may become conditionally essential in certain situations, including intensive athletic training or certain gastrointestinal disorders...
, than it is to be mutated into a hydrophobic residue such as leucine
Leucine
Leucine is a branched-chain α-amino acid with the chemical formula HO2CCHCH2CH2. Leucine is classified as a hydrophobic amino acid due to its aliphatic isobutyl side chain. It is encoded by six codons and is a major component of the subunits in ferritin, astacin and other 'buffer' proteins...
. This is primarily due to redundancy in the genetic code
Genetic code
The genetic code is the set of rules by which information encoded in genetic material is translated into proteins by living cells....
, which translates similar codons into similar amino acids. Furthermore, mutating an amino acid to a residue with significantly different properties could affect the folding
Protein folding
Protein folding is the process by which a protein structure assumes its functional shape or conformation. It is the physical process by which a polypeptide folds into its characteristic and functional three-dimensional structure from random coil....
and/or activity of the protein. There is therefore usually strong selective pressure to remove such mutations quickly from a population.
If we have two amino acid sequences in front of us, we should be able to say something about how likely they are to be derived from a common ancestor, or homologous
Homology (biology)
Homology forms the basis of organization for comparative biology. In 1843, Richard Owen defined homology as "the same organ in different animals under every variety of form and function". Organs as different as a bat's wing, a seal's flipper, a cat's paw and a human hand have a common underlying...
. If we can line up the two sequences using a sequence alignment
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
algorithm such that the mutations required to transform a hypothetical ancestor sequence into both of the current sequences would be evolutionarily plausible, then we'd like to assign a high score to the comparison of the sequences.
To this end, we will construct a 20x20 matrix where the
th entry is equal to the probability of the th amino acid being transformed into the th amino acid in a certain amount of evolutionary time. There are many different ways to construct such a matrix, called a substitution matrix. Here are the most commonly used ones:Identity matrix
The simplest possible substitution matrix would be one in which each amino acid is considered maximally similar to itself, but not able to transform into any other amino acid. This matrix would look like:This identity matrix
Identity matrix
In linear algebra, the identity matrix or unit matrix of size n is the n×n square matrix with ones on the main diagonal and zeros elsewhere. It is denoted by In, or simply by I if the size is immaterial or can be trivially determined by the context...
will succeed in the alignment of very similar amino acid sequences but will be miserable at aligning two distantly related sequences. We need to figure out all the probabilities in a more rigorous fashion. It turns out that an empirical examination of previously aligned sequences works best.
Log-odds matrices
We express the probabilitiesProbability
Probability is ordinarily used to describe an attitude of mind towards some proposition of whose truth we arenot certain. The proposition of interest is usually of the form "Will a specific event occur?" The attitude of mind is of the form "How certain are we that the event will occur?" The...
of transformation in what are called log-odds score
Score (statistics)
In statistics, the score, score function, efficient score or informant plays an important role in several aspects of inference...
s. The scores matrix S is defined as
where
is the probability that amino acid transforms into amino acid and is the frequency of amino acid i. The base of the logarithm is not important, and you will often see the same substitution matrix expressed in different bases.PAM
One of the first amino acid substitution matrices, the PAM (Point Accepted MutationPoint accepted mutation
Point accepted mutation , is a set of matrices used to score sequence alignments. The PAM matrices were introduced by Margaret Dayhoff in 1978 based on 1572 observed mutations in 71 families of closely related proteins...
) matrix was developed by Margaret Dayhoff
Margaret Oakley Dayhoff
Dr. Margaret Belle Dayhoff was an American physical chemist and a pioneer in the field of bioinformatics...
in the 1970s. This matrix is calculated by observing the differences in closely related proteins. The PAM1 matrix estimates what rate of substitution would be expected if 1% of the amino acids had changed. The PAM1 matrix is used as the basis for calculating other matrices by assuming that repeated mutations would follow the same pattern as those in the PAM1 matrix, and multiple substitutions can occur at the same site. Using this logic, Dayhoff derived matrices as high as PAM250. Usually the PAM 30 and the PAM70 are used.
A matrix for divergent sequences can be calculated from a matrix for closely related sequences by taking the second matrix to a power. For instance, we can roughly approximate the WIKI2 matrix from the WIKI1 matrix by saying
where is WIKI1 and is WIKI2. This is how the PAM250 matrix is calculated.BLOSUM
Dayhoff's methodology of comparing closely related species turned out not to work very well for aligning evolutionarily divergent sequences. Sequence changes over long evolutionary time scales are not well approximated by compounding small changes that occur over short time scales. The BLOSUMBLOSUM
The BLOSUM matrix is a substitution matrix used for sequence alignment of proteins. BLOSUM matrices are used to score alignments between evolutionarily divergent protein sequences. They are based on local alignments. BLOSUM matrices were first introduced in a paper by Henikoff and Henikoff...
(BLOck SUbstitution Matrix) series of matrices rectifies this problem. Henikoff and Henikoff constructed these matrices using multiple alignments of evolutionarily divergent proteins. The probabilities used in the matrix calculation are computed by looking at "blocks" of conserved sequences found in multiple protein alignments. These conserved sequences are assumed to be of functional importance within related proteins. To reduce bias from closely related sequences, segments in a block with a sequence identity above a certain threshold were clustered giving weight to each such cluster (Henikoff and Henikoff). For the BLOSUM62 matrix, this threshold was set at 62%. Pairs frequencies were then counted between clusters, hence pairs were only counted between segments less than 62% identical. One would use a higher numbered BLOSUM matrix for aligning two closely related sequences and a lower number for more divergent sequences.
It turns out that the BLOSUM62 matrix does an excellent job detecting similarities in distant sequences, and this is the matrix used by default in most recent alignment applications such as BLAST
BLAST
In bioinformatics, Basic Local Alignment Search Tool, or BLAST, is an algorithm for comparing primary biological sequence information, such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences...
.
Differences between PAM and BLOSUM
- PAM matrices are based on an explicit evolutionary model (i.e. replacements are counted on the branches of a phylogenetic tree), whereas the BLOSUM matrices are based on an implicit model of evolution.
- The PAM matrices are based on mutations observed throughout a global alignment, this includes both highly conserved and highly mutable regions. The BLOSUM matrices are based only on highly conserved regions in series of alignments forbidden to contain gaps.
- The method used to count the replacements is different: unlike the PAM matrix, the BLOSUM procedure uses groups of sequences within which not all mutations are counted the same.
- Higher numbers in the PAM matrix naming scheme denote larger evolutionary distance, while larger numbers in the BLOSUM matrix naming scheme denote higher sequence similarity and therefore smaller evolutionary distance. Example: PAM150 is used for more distant sequences than PAM100; BLOSUM62 is used for closer sequences than Blosum50.
Extensions and improvements
Many specialized substitution matrices have been developed that describe the amino acid substitution rates in specific structural or sequence contexts, such as in transmembrane alpha helices, for combinations of secondary structure states and solvent accessibility states, or for local sequence-structure contexts. These context-specific substitution matrices lead to generally improved alignment quality at some cost of speed but are not yet widely used. Recently, sequence context-specific amino acid similarities have been derived that do not need substitution matrices but that rely on a library of sequence contexts instead. Using this idea, a context-specific extension of the popular BLASTBLAST
In bioinformatics, Basic Local Alignment Search Tool, or BLAST, is an algorithm for comparing primary biological sequence information, such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences...
program has been demonstrated to achieve a twofold sensitivity improvement for remotely related sequences over BLAST at similar speeds (CS-BLAST
CS-BLAST
CS-BLAST , an improved version of BLAST , is a program for protein sequence searching....
).
Terminology
Although "transition matrix" is often used interchangeably with "substitution matrix" in fields other than bioinformatics, the former term is problematic in bioinformatics. With regards to nucleotide substitutions, "transitionTransition (genetics)
In genetics, a transition is a point mutation that changes a purine nucleotide to another purine or a pyrimidine nucleotide to another pyrimidine . Approximately two out of three single nucleotide polymorphisms are transitions....
" is also used to indicate those substitutions that are between the two-ring purine
Purine
A purine is a heterocyclic aromatic organic compound, consisting of a pyrimidine ring fused to an imidazole ring. Purines, including substituted purines and their tautomers, are the most widely distributed kind of nitrogen-containing heterocycle in nature....
s (A → G and G → A) or are between the one-ring pyrimidine
Pyrimidine
Pyrimidine is a heterocyclic aromatic organic compound similar to benzene and pyridine, containing two nitrogen atoms at positions 1 and 3 of the six-member ring...
s (C → T and T → C). Because these substitutions do not require a change in the number of rings, they occur more frequently than the other substitutions. "Transversion
Transversion
In molecular biology, transversion refers to the substitution of a purine for a pyrimidine or vice versa. It can only be reverted by a spontaneous reversion. Because this type of mutation changes the chemical structure dramatically, the consequences of this change tend to be more drastic than those...
" is the term used to indicate the slower-rate substitutions that change a purine to a pyrimidine or vice versa (A ↔ C, A ↔ T, G ↔ C, and G ↔ T).