

Human genetic clustering
Encyclopedia
Human genetic clustering analysis uses mathematical cluster analysis of the degree of similarity of genetic data between individuals and groups to infer population structures and assign individuals to groups that often correspond with their self-identified geographical ancestry. A similar analysis can be done using principal components analysis
, which in earlier research was a popular method. Many of recent studies in the past few years have returned to using principal components analysis.
Phylogenetic tree
Neighbor-joining
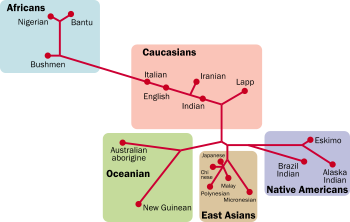
Clusters by Rosenberg
In 2004, Lynn Jorde and Steven Wooding argued that "Analysis of many loci now yields reasonably accurate estimates of genetic similarity among individuals, rather than populations. Clustering of individuals is correlated with geographic origin or ancestry."

 A study by Neil Risch
A study by Neil Risch
in 2005 used 326 microsatellite markers and self-identified race/ethnic group (SIRE), white (European American), African-American (black), Asian and Hispanic (individuals involved in the study had to choose from one of these categories), to representing discrete "populations", and showed distinct and non-overlapping clustering of the white, African-American and Asian samples. The results were claimed to confirm the integrity of self-described ancestry: "We have shown a nearly perfect correspondence between genetic cluster and SIRE for major ethnic groups living in the United States, with a discrepancy rate of only 0.14%."(Tang, 2005)
Studies such as those by Risch and Rosenberg
use a computer program called STRUCTURE to find human populations (gene clusters). It is a statistical program that works by placing individuals into one of two clusters based on their overall genetic similarity, many possible pairs of clusters are tested per individual to generate multiple clusters. These populations are based on multiple genetic markers that are often shared between different human populations even over large geographic ranges. The notion of a genetic cluster is that people within the cluster share on average similar allele frequencies to each other than to those in other clusters. (A. W. F. Edwards
, 2003 but see also infobox "Multi Locus Allele Clusters") In a test of idealised populations, the computer programme STRUCTURE was found to consistently under-estimate the numbers of populations in the data set when high migration rates between populations and slow mutation rates (such as single-nucleotide polymorphisms) were considered.
Nevertheless the Rosenberg et al. (2002) paper shows that individuals can be assigned to specific clusters to a high degree of accuracy. One of the underlying questions regarding the distribution of human genetic diversity is related to the degree to which genes are shared between the observed clusters. It has been observed repeatedly that the majority of variation observed in the global human population is found within populations. This variation is usually calculated using Sewall Wright
's Fixation index (FST), which is an estimate of between to within group variation. The degree of human genetic variation is a little different depending upon the gene type studied, but in general it is common to claim that ~85% of genetic variation is found within groups, ~6–10% between groups within the same continent and ~6–10% is found between continental groups. For example The Human Genome Project states "two random individuals from any one group are almost as different [genetically] as any two random individuals from the entire world." Sarich and Miele, however, have argued that estimates of genetic difference between individuals of different populations fail to take into account human diploidity.
Additionally, Edwards (2003) claims in his essay "Lewontin's Fallacy
" that: "It is not true, as Nature claimed, that 'two random individuals from any one group are almost as different as any two random individuals from the entire world'" and Risch et al. (2002) state "Two Caucasians are more similar to each other genetically than a Caucasian and an Asian." It should be noted that these statements are not the same. Risch et al. simply state that two indigenous
individuals from the same geographical region are more similar to each other than either is to an indigenous individual from a different geographical region, a claim few would argue with. Jorde et al. put it like this:
Whereas Edwards claims that it is not true that the differences between individuals from different geographical regions represent only a small proportion of the variation within the human population (he claims that within group differences between individuals are not almost as large as between group differences). Bamshad et al. (2004) used the data from Rosenberg et al. (2002) to investigate the extent of genetic differences between individuals within continental groups relative to genetic differences between individuals between continental groups. They found that though these individuals could be classified very accurately to continental clusters, there was a significant degree of genetic overlap on the individual level, to the extent that, using 377 loci, individual Europeans were about 38% of the time more genetically similar to East Asians than to other Europeans.
The existence of allelic clines and the observation that the bulk of human variation is continuously distributed, has led some scientists to conclude that any categorization schema attempting to partition that variation meaningfully will necessarily create artificial truncations. (Kittles & Weiss 2003). It is for this reason, Reanne Frank argues, that attempts to allocate individuals into ancestry groupings based on genetic information have yielded varying results that are highly dependent on methodological design. Serre and Pääbo (2004) make a similar claim:
In a response to Serre and Pääbo (2004), Rosenberg et al. (2005) make three relevant observations. Firstly they maintain that their clustering analysis is robust. Secondly they agree with Serre and Pääbo that membership of multiple clusters can be interpreted as evidence for clinality (isolation by distance), though they also comment that this may also be due to admixture between neighbouring groups (small island model). Thirdly they comment that evidence of clusterdness is not evidence for any concepts of "biological race".
Risch et al. (2002) state that "two Caucasians are more similar to each other genetically than a Caucasian and an Asian", but Bamshad et al. (2004) used the same data set as Rosenberg et al. (2002) to show that Europeans are more similar to Asians 38% of the time than they are to other Europeans when only 377 microsatellite markers are analysed.
In agreement with the observation of Bamshad et al. (2004), Witherspoon et al. (2007) have shown that many more than 326 or 377 microsatellite loci are required in order to show that individuals are always more similar to individuals in their own population group than to individuals in different population groups, even for three distinct populations.
Witherspoon et al. (2007) have argued that even when individuals can be reliably assigned to specific population groups, it may still be possible for two randomly chosen individuals from different populations/clusters to be more similar to each other than to a randomly chosen member of their own cluster. They found that many thousands of genetic markers had to be used in order for the answer to the question "How often is a pair of individuals from one population genetically more dissimilar than two individuals chosen from two different populations?" to be "never". This assumed three population groups separated by large geographic ranges (European, African and East Asian). The entire world population is much more complex and studying an increasing number of groups would require an increasing number of markers for the same answer. Witherspoon et al. conclude that "caution should be used when using geographic or genetic ancestry to make inferences about individual phenotypes."
Clustering does not particularly correspond to continental divisions. Depending on the parameters given to their analytical program, Rosenberg and Pritchard were able to construct between divisions of between 4 and 20 clusters of the genomes studied, although they excluded analysis with more than 6 clusters from their published article. Probability values for various cluster configurations varied widely, with the single most likely configuration coming with 16 clusters although other 16-cluster configurations had low probabilities. Overall, "there is no clear evidence that K=6 was the best estimate" according to geneticist Deborah Bolnick (2008:76-77).
A study by the The HUGO Pan-Asian SNP Consortium in 2009 using the similar principle components analysis found that East Asian and South-East Asian populations clustered together, and suggested a common origin for these populations. At the same time they observed a broad discontinuity between this cluster and South Asia, commenting "most of the Indian populations showed evidence of shared ancestry with European populations". It was noted that "genetic ancestry is strongly correlated with linguistic affiliations as well as geography".
Principal components analysis
Principal component analysis is a mathematical procedure that uses an orthogonal transformation to convert a set of observations of possibly correlated variables into a set of values of uncorrelated variables called principal components. The number of principal components is less than or equal to...
, which in earlier research was a popular method. Many of recent studies in the past few years have returned to using principal components analysis.
Phylogenetic treePhylogenetic treeA phylogenetic tree or evolutionary tree is a branching diagram or "tree" showing the inferred evolutionary relationships among various biological species or other entities based upon similarities and differences in their physical and/or genetic characteristics...
by Cavalli-Sforza & al. (1994)
Neighbor-joiningNeighbor-joiningIn bioinformatics, neighbor joining is a bottom-up clustering method for the creation of phenetic trees , created by Naruya Saitou and Masatoshi Nei...
method, by Naruya Saitou and Masatoshi NeiMasatoshi Neiis Evan Pugh Professor of Biology at Pennsylvania State University and Director of the since 1990. He was born in 1931 in Miyazaki Prefecture, on Kyūshū Island, Japan...
(2002)
Clusters by RosenbergNoah RosenbergNoah Rosenberg is a geneticist working in evolutionary biology, human genetics, and population genetics, now Associate Professor at Stanford University...
& al. (2006)
In 2004, Lynn Jorde and Steven Wooding argued that "Analysis of many loci now yields reasonably accurate estimates of genetic similarity among individuals, rather than populations. Clustering of individuals is correlated with geographic origin or ancestry."Neil Risch
Neil Risch is an American human geneticist and professor at the University of California, San Francisco . Risch is the Lamond Family Foundation Distinguished Professor in Human Genetics and Director of the Institute for Human Genetics and Professor of Epidemiology and Biostatistics at UCSF.Known...
in 2005 used 326 microsatellite markers and self-identified race/ethnic group (SIRE), white (European American), African-American (black), Asian and Hispanic (individuals involved in the study had to choose from one of these categories), to representing discrete "populations", and showed distinct and non-overlapping clustering of the white, African-American and Asian samples. The results were claimed to confirm the integrity of self-described ancestry: "We have shown a nearly perfect correspondence between genetic cluster and SIRE for major ethnic groups living in the United States, with a discrepancy rate of only 0.14%."(Tang, 2005)
Studies such as those by Risch and Rosenberg
Noah Rosenberg
Noah Rosenberg is a geneticist working in evolutionary biology, human genetics, and population genetics, now Associate Professor at Stanford University...
use a computer program called STRUCTURE to find human populations (gene clusters). It is a statistical program that works by placing individuals into one of two clusters based on their overall genetic similarity, many possible pairs of clusters are tested per individual to generate multiple clusters. These populations are based on multiple genetic markers that are often shared between different human populations even over large geographic ranges. The notion of a genetic cluster is that people within the cluster share on average similar allele frequencies to each other than to those in other clusters. (A. W. F. Edwards
A. W. F. Edwards
Anthony William Fairbank Edwards is a British statistician, geneticist, and evolutionary biologist, sometimes called Fisher's Edwards. He is a Life Fellow of Gonville and Caius College and retired Professor of Biometry at the University of Cambridge, and holds both the ScD and LittD degrees. A...
, 2003 but see also infobox "Multi Locus Allele Clusters") In a test of idealised populations, the computer programme STRUCTURE was found to consistently under-estimate the numbers of populations in the data set when high migration rates between populations and slow mutation rates (such as single-nucleotide polymorphisms) were considered.
Nevertheless the Rosenberg et al. (2002) paper shows that individuals can be assigned to specific clusters to a high degree of accuracy. One of the underlying questions regarding the distribution of human genetic diversity is related to the degree to which genes are shared between the observed clusters. It has been observed repeatedly that the majority of variation observed in the global human population is found within populations. This variation is usually calculated using Sewall Wright
Sewall Wright
Sewall Green Wright was an American geneticist known for his influential work on evolutionary theory and also for his work on path analysis. With R. A. Fisher and J.B.S. Haldane, he was a founder of theoretical population genetics. He is the discoverer of the inbreeding coefficient and of...
's Fixation index (FST), which is an estimate of between to within group variation. The degree of human genetic variation is a little different depending upon the gene type studied, but in general it is common to claim that ~85% of genetic variation is found within groups, ~6–10% between groups within the same continent and ~6–10% is found between continental groups. For example The Human Genome Project states "two random individuals from any one group are almost as different [genetically] as any two random individuals from the entire world." Sarich and Miele, however, have argued that estimates of genetic difference between individuals of different populations fail to take into account human diploidity.
Additionally, Edwards (2003) claims in his essay "Lewontin's Fallacy
Lewontin's Fallacy
Human genetic diversity: Lewontin's fallacy is a 2003 paper by A. W. F. Edwards that refers to an argument first made by Richard Lewontin in his 1972 article The apportionment of human diversity, which argued that race for humans is not a valid taxonomic construct. Edwards' paper criticized and...
" that: "It is not true, as Nature claimed, that 'two random individuals from any one group are almost as different as any two random individuals from the entire world'" and Risch et al. (2002) state "Two Caucasians are more similar to each other genetically than a Caucasian and an Asian." It should be noted that these statements are not the same. Risch et al. simply state that two indigenous
Indigenous peoples
Indigenous peoples are ethnic groups that are defined as indigenous according to one of the various definitions of the term, there is no universally accepted definition but most of which carry connotations of being the "original inhabitants" of a territory....
individuals from the same geographical region are more similar to each other than either is to an indigenous individual from a different geographical region, a claim few would argue with. Jorde et al. put it like this:
Whereas Edwards claims that it is not true that the differences between individuals from different geographical regions represent only a small proportion of the variation within the human population (he claims that within group differences between individuals are not almost as large as between group differences). Bamshad et al. (2004) used the data from Rosenberg et al. (2002) to investigate the extent of genetic differences between individuals within continental groups relative to genetic differences between individuals between continental groups. They found that though these individuals could be classified very accurately to continental clusters, there was a significant degree of genetic overlap on the individual level, to the extent that, using 377 loci, individual Europeans were about 38% of the time more genetically similar to East Asians than to other Europeans.
The existence of allelic clines and the observation that the bulk of human variation is continuously distributed, has led some scientists to conclude that any categorization schema attempting to partition that variation meaningfully will necessarily create artificial truncations. (Kittles & Weiss 2003). It is for this reason, Reanne Frank argues, that attempts to allocate individuals into ancestry groupings based on genetic information have yielded varying results that are highly dependent on methodological design. Serre and Pääbo (2004) make a similar claim:
In a response to Serre and Pääbo (2004), Rosenberg et al. (2005) make three relevant observations. Firstly they maintain that their clustering analysis is robust. Secondly they agree with Serre and Pääbo that membership of multiple clusters can be interpreted as evidence for clinality (isolation by distance), though they also comment that this may also be due to admixture between neighbouring groups (small island model). Thirdly they comment that evidence of clusterdness is not evidence for any concepts of "biological race".
Risch et al. (2002) state that "two Caucasians are more similar to each other genetically than a Caucasian and an Asian", but Bamshad et al. (2004) used the same data set as Rosenberg et al. (2002) to show that Europeans are more similar to Asians 38% of the time than they are to other Europeans when only 377 microsatellite markers are analysed.
| x | Africans | Europeans | Asians |
|---|---|---|---|
| Europeans | 36.5 | — | — |
| Asians | 35.5 | 38.3 | — |
| Indigenous Americans | 26.1 | 33.4 | 35 |
In agreement with the observation of Bamshad et al. (2004), Witherspoon et al. (2007) have shown that many more than 326 or 377 microsatellite loci are required in order to show that individuals are always more similar to individuals in their own population group than to individuals in different population groups, even for three distinct populations.
Witherspoon et al. (2007) have argued that even when individuals can be reliably assigned to specific population groups, it may still be possible for two randomly chosen individuals from different populations/clusters to be more similar to each other than to a randomly chosen member of their own cluster. They found that many thousands of genetic markers had to be used in order for the answer to the question "How often is a pair of individuals from one population genetically more dissimilar than two individuals chosen from two different populations?" to be "never". This assumed three population groups separated by large geographic ranges (European, African and East Asian). The entire world population is much more complex and studying an increasing number of groups would require an increasing number of markers for the same answer. Witherspoon et al. conclude that "caution should be used when using geographic or genetic ancestry to make inferences about individual phenotypes."
Clustering does not particularly correspond to continental divisions. Depending on the parameters given to their analytical program, Rosenberg and Pritchard were able to construct between divisions of between 4 and 20 clusters of the genomes studied, although they excluded analysis with more than 6 clusters from their published article. Probability values for various cluster configurations varied widely, with the single most likely configuration coming with 16 clusters although other 16-cluster configurations had low probabilities. Overall, "there is no clear evidence that K=6 was the best estimate" according to geneticist Deborah Bolnick (2008:76-77).
A study by the The HUGO Pan-Asian SNP Consortium in 2009 using the similar principle components analysis found that East Asian and South-East Asian populations clustered together, and suggested a common origin for these populations. At the same time they observed a broad discontinuity between this cluster and South Asia, commenting "most of the Indian populations showed evidence of shared ancestry with European populations". It was noted that "genetic ancestry is strongly correlated with linguistic affiliations as well as geography".
See also
- Human genetic variationHuman genetic variationHuman genetic variation refers to genetic differences both within and among populations. There may be multiple variants of any given gene in the human population , leading to polymorphism. Many genes are not polymorphic, meaning that only a single allele is present in the population: that allele is...
- Population groups in biomedicine
- Y-DNA haplogroups by ethnic groupsY-DNA haplogroups by ethnic groupsListed here are notable ethnic groups by Y-DNA haplogroups based on relevant studies. The data is presented in two columns for each haplogroup with the first being the sample size and the second the percentage in the haplogroup designated by the column header...