

Neighbor-joining
Encyclopedia
In bioinformatics
, neighbor joining is a bottom-up clustering method for the creation of phenetic trees (phenograms), created by Naruya Saitou and Masatoshi Nei
. Usually used for trees based on DNA
or protein
sequence
data, the algorithm requires knowledge of the distance between each pair of taxa (e.g., species or sequences) to form the tree.
, and iterates over the following steps until the tree is completely resolved and all branch lengths are known:
 taxa, calculate
taxa, calculate 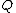 as follows:
as follows:
where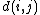 is the distance between taxa
is the distance between taxa 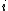 and
and 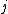 .
.
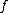 and
and 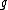 are the paired taxa and
are the paired taxa and  is the newly generated node.):
is the newly generated node.):
and, by reflection:
where is the new node,
is the new node, 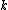 is the node for which we want to calculate the distance and
is the node for which we want to calculate the distance and 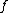 and
and 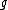 are the members of the pair just joined.
are the members of the pair just joined.
 taxa takes requires
taxa takes requires  iterations, and since at each step one has to build and search
iterations, and since at each step one has to build and search 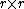 matrices, the algorithm can be implemented so as to obtain a time complexity of
matrices, the algorithm can be implemented so as to obtain a time complexity of 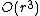 .
.
We obtain the following values for the Q matrix:
In the example above, two pairs of taxa have the lowest value, namely −40. We can select either of them for the second step of the algorithm. We follow the example assuming that we joined taxa A and B together.
If denotes the new node, then the branch lengths of edges
denotes the new node, then the branch lengths of edges 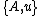 and
and 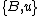 are respectively 6 and 1, by the above formula.
are respectively 6 and 1, by the above formula.
We then proceed to updating the distance matrix, by computing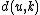 according to the above formula for every node
according to the above formula for every node 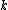 . In this case, we obtain
. In this case, we obtain 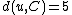 and
and 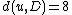 . The resulting distance matrix is:
. The resulting distance matrix is:
We can start the procedure anew taking this matrix as the original distance matrix. In our example, it suffices to do one more step of the recursion to obtain the complete tree.
that constructs the tree in a step-wise fashion. Even though it is sub-optimal in this sense, it has been extensively tested and usually finds a tree that is quite close to the optimal tree. Nevertheless, it has been largely superseded by phylogenetic methods that do not rely on distance measures and offer superior accuracy under most conditions.
The main virtue of neighbor joining relative to these other methods is its computational efficiency. That is, neighbor joining is a polynomial-time algorithm. It can be used on very large data sets for which other means of analysis (e.g. minimum evolution, maximum parsimony
, maximum likelihood
) are computation
ally prohibitive. Unlike the UPGMA
algorithm for tree reconstruction, neighbor joining does not assume that all lineages evolve at the same rate (molecular clock hypothesis) and produces an unrooted tree. Rooted trees can be created by using an outgroup
and the root can then effectively be placed on the point in the tree where the edge from the outgroup connects.
Furthermore, neighbor joining is statistically consistent under many models of evolution. Hence, given data of sufficient length, neighbor joining will reconstruct the true tree with high probability.
Atteson proved that if each entry in the distance matrix differs from the true distance by less than half of the shortest branch length in the tree,
then neighbor joining will construct the correct tree.
RapidNJ and
NINJA
are fast implementations of the neighbor joining algorithm.
Although neighbor joining relies on a matrix whose entries are natural
, some branch lengths in the resulting topology may be real numbers.
Bioinformatics
Bioinformatics is the application of computer science and information technology to the field of biology and medicine. Bioinformatics deals with algorithms, databases and information systems, web technologies, artificial intelligence and soft computing, information and computation theory, software...
, neighbor joining is a bottom-up clustering method for the creation of phenetic trees (phenograms), created by Naruya Saitou and Masatoshi Nei
Masatoshi Nei
is Evan Pugh Professor of Biology at Pennsylvania State University and Director of the since 1990. He was born in 1931 in Miyazaki Prefecture, on Kyūshū Island, Japan...
. Usually used for trees based on DNA
DNA
Deoxyribonucleic acid is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms . The DNA segments that carry this genetic information are called genes, but other DNA sequences have structural purposes, or are involved in...
or protein
Protein
Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form, facilitating a biological function. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of...
sequence
Primary structure
The primary structure of peptides and proteins refers to the linear sequence of its amino acid structural units. The term "primary structure" was first coined by Linderstrøm-Lang in 1951...
data, the algorithm requires knowledge of the distance between each pair of taxa (e.g., species or sequences) to form the tree.
The algorithm
Neighbor joining starts with a completely unresolved tree, whose topology corresponds to that of a star networkStar network
Star networks are one of the most common computer network topologies. In its simplest form, a star network consists of one central switch, hub or computer, which acts as a conduit to transmit messages...
, and iterates over the following steps until the tree is completely resolved and all branch lengths are known:
- Based on the current distance matrixDistance matrixIn mathematics, computer science and graph theory, a distance matrix is a matrix containing the distances, taken pairwise, of a set of points...
calculate the matrix (defined below).
(defined below). - Find the pair of taxa in
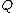 with the lowest value. Create a node on the tree that joins these two taxa (i.e., join the closest neighbors, as the algorithm name implies).
with the lowest value. Create a node on the tree that joins these two taxa (i.e., join the closest neighbors, as the algorithm name implies). - Calculate the distance of each of the taxaTaxon|thumb|270px|[[African elephants]] form a widely-accepted taxon, the [[genus]] LoxodontaA taxon is a group of organisms, which a taxonomist adjudges to be a unit. Usually a taxon is given a name and a rank, although neither is a requirement...
in the pair to this new node. - Calculate the distance of all taxa outside of this pair to the new node.
- Start the algorithm again, considering the pair of joined neighbors as a single taxon and using the distances calculated in the previous step.
The Q-matrix
Based on a distance matrix relating the taxa, calculate as follows:
where
is the distance between taxa and .Distance of the pair members to the new node
For each neighbor in the pair just joined, use the following formula to calculate the distance to the new node. (Taxa and are the paired taxa and is the newly generated node.):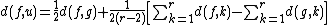
and, by reflection:
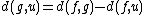
Distance of the other taxa to the new node
For each taxon not considered in the previous step, we calculate the distance to the new node as follows: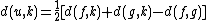
where
is the new node, is the node for which we want to calculate the distance and and are the members of the pair just joined.Complexity
Since neighbor joining on a set of taxa takes requires iterations, and since at each step one has to build and search matrices, the algorithm can be implemented so as to obtain a time complexity of .Example
Let us assume that we have four taxa (A, B, C, D) and the following distance matrix:| A | B | C | D | |
|---|---|---|---|---|
| A | 0 | 7 | 11 | 14 |
| B | 7 | 0 | 6 | 9 |
| C | 11 | 6 | 0 | 7 |
| D | 14 | 9 | 7 | 0 |
We obtain the following values for the Q matrix:
| A | B | C | D | |
|---|---|---|---|---|
| A | 0 | −40 | −34 | −34 |
| B | −40 | 0 | −34 | −34 |
| C | −34 | −34 | 0 | −40 |
| D | −34 | −34 | −40 | 0 |
In the example above, two pairs of taxa have the lowest value, namely −40. We can select either of them for the second step of the algorithm. We follow the example assuming that we joined taxa A and B together.
If
denotes the new node, then the branch lengths of edges and are respectively 6 and 1, by the above formula.We then proceed to updating the distance matrix, by computing
according to the above formula for every node . In this case, we obtain and . The resulting distance matrix is:| AB | C | D | |
|---|---|---|---|
| AB | 0 | 5 | 8 |
| C | 5 | 0 | 7 |
| D | 8 | 7 | 0 |
We can start the procedure anew taking this matrix as the original distance matrix. In our example, it suffices to do one more step of the recursion to obtain the complete tree.
Advantages and disadvantages
Neighbor joining is based on the minimum-evolution criterion, i.e. the topology that gives the least total branch length is preferred at each step of the algorithm. However, neighbor joining may not find the true tree topology with least total branch length because it is a greedy algorithmGreedy algorithm
A greedy algorithm is any algorithm that follows the problem solving heuristic of making the locally optimal choice at each stagewith the hope of finding the global optimum....
that constructs the tree in a step-wise fashion. Even though it is sub-optimal in this sense, it has been extensively tested and usually finds a tree that is quite close to the optimal tree. Nevertheless, it has been largely superseded by phylogenetic methods that do not rely on distance measures and offer superior accuracy under most conditions.
The main virtue of neighbor joining relative to these other methods is its computational efficiency. That is, neighbor joining is a polynomial-time algorithm. It can be used on very large data sets for which other means of analysis (e.g. minimum evolution, maximum parsimony
Maximum parsimony
Parsimony is a non-parametric statistical method commonly used in computational phylogenetics for estimating phylogenies. Under parsimony, the preferred phylogenetic tree is the tree that requires the least evolutionary change to explain some observed data....
, maximum likelihood
Maximum likelihood
In statistics, maximum-likelihood estimation is a method of estimating the parameters of a statistical model. When applied to a data set and given a statistical model, maximum-likelihood estimation provides estimates for the model's parameters....
) are computation
Computation
Computation is defined as any type of calculation. Also defined as use of computer technology in Information processing.Computation is a process following a well-defined model understood and expressed in an algorithm, protocol, network topology, etc...
ally prohibitive. Unlike the UPGMA
UPGMA
UPGMA is a simple agglomerative or hierarchical clustering method used in bioinformatics for the creation of phenetic trees...
algorithm for tree reconstruction, neighbor joining does not assume that all lineages evolve at the same rate (molecular clock hypothesis) and produces an unrooted tree. Rooted trees can be created by using an outgroup
Outgroup
In cladistics or phylogenetics, an outgroup is a group of organisms that serves as a reference group for determination of the evolutionary relationship among three or more monophyletic groups of organisms....
and the root can then effectively be placed on the point in the tree where the edge from the outgroup connects.
Furthermore, neighbor joining is statistically consistent under many models of evolution. Hence, given data of sufficient length, neighbor joining will reconstruct the true tree with high probability.
Atteson proved that if each entry in the distance matrix differs from the true distance by less than half of the shortest branch length in the tree,
then neighbor joining will construct the correct tree.
RapidNJ and
NINJA
are fast implementations of the neighbor joining algorithm.
Although neighbor joining relies on a matrix whose entries are natural
Natural number
In mathematics, the natural numbers are the ordinary whole numbers used for counting and ordering . These purposes are related to the linguistic notions of cardinal and ordinal numbers, respectively...
, some branch lengths in the resulting topology may be real numbers.
External links
- The Neighbor-Joining Method — a tutorial