

Protein nuclear magnetic resonance spectroscopy
Encyclopedia
Nuclear magnetic resonance spectroscopy of proteins (usually abbreviated protein NMR) is a field of structural biology
in which NMR spectroscopy
is used to obtain information about the structure and dynamics of proteins. The field was pioneered by Richard R. Ernst
and Kurt Wüthrich
http://www.nature.com/nsmb/journal/v8/n11/full/nsb1101-923.html, among others. Protein NMR techniques are continually being used and improved in both academia
and the biotech industry
. Structure determination by NMR spectroscopy usually consists of several following phases, each using a separate set of highly specialized techniques. The sample is prepared, resonances are assigned, restraints are generated and a structure is calculated and validated.
 Protein nuclear magnetic resonance is performed on aqueous samples of highly purified
Protein nuclear magnetic resonance is performed on aqueous samples of highly purified
protein. Usually the sample consist of between 300 and 600 microlitres with a protein concentration in the range 0.1 – 3 millimolar
. The source of the protein can be either natural or produced in an expression system
using recombinant DNA
techniques through genetic engineering
. Recombinantly expressed
proteins are usually easier to produce in sufficient quantity, and makes isotopic
labelling
possible.
The purified protein is usually dissolved in a buffer solution
and adjusted to the desired solvent conditions. The NMR sample is prepared in a thin walled glass tube
.
by which it can be recognized. However, in large molecules such as proteins the number of resonances can typically be several thousand and a one-dimensional spectrum inevitably has incidental overlaps. Therefore multidimensional experiments are performed which correlate the frequencies of distinct nuclei. The additional dimensions decrease the chance of overlap and have a larger information content since they correlate signals from nuclei within a specific part of the molecule. Magnetization is transferred into the sample using pulses of electromagnetic (radiofrequency) energy and between nuclei using delays; the process is described with so-called pulse sequence
s. Pulse sequences allow the experimenter to investigate and select specific types of connections between nuclei. The array of nuclear magnetic resonance experiments used on proteins fall in two main categories — one where magnetization is transferred through the chemical bonds, and one where the transfer is through space, irrespective of the bonding structure. The first category is used to assign the different chemical shift
s to a specific nucleus, and the second is primarily used to generate the distance restraints used in the structure calculation, and in the assignment with unlabelled protein.
Depending on the concentration of the sample, on the magnetic field of the spectrometer, and on the type of experiment, a single multidimensional nuclear magnetic resonance experiment on a protein sample may take hours or even several days to obtain suitable signal-to-noise ratio through signal averaging, and to allow for sufficient evolution of magnetization transfer through the various dimensions of the experiment. Other things being equal, higher-dimensional experiments will take longer than lower-dimensional experiments.
Typically the first experiment to be measured with an isotope-labelled protein is a 2D heteronuclear single quantum correlation
(HSQC) spectrum where "heteronuclear" refers to nuclei other than 1H. In theory the heteronuclear single quantum correlation has one peak for each H bound to a heteronucleus. Thus in the 15N-HSQC one signal is expected for each amino acid residue with the exception of proline
which has no amide-hydrogen due to the cyclic nature of its backbone. Tryptophan
and certain other residues with N-containing sidechains also give rise to additional signals. The 15N-HSQC is often referred to as the fingerprint of a protein because each protein has a unique pattern of signal positions. Analysis of the 15N-HSQC allows researchers to evaluate whether the expected number of peaks is present and thus to identify possible problems due to multiple conformations or sample heterogeneity. The relatively quick heteronuclear single quantum correlation experiment helps determine the feasibility of doing subsequent longer, more expensive, and more elaborate experiments. It is not possible to assign peaks to specific atoms from the heteronuclear single quantum correlation alone.
corresponds to which atom. This is typically achieved by sequential walking
using information derived from several different types of NMR experiment. The exact procedure depends on whether the protein is isotopically labelled
or not, since a lot of the assignment experiments depend on carbon-13 and nitrogen-15.
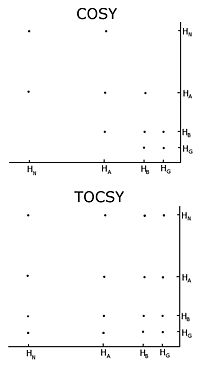
(COSY), of which several types include conventional correlation spectroscopy, total correlation spectroscopy (TOCSY) and nuclear Overhauser effect
spectroscopy (NOESY). A two-dimensional nuclear magnetic resonance experiment produces a two-dimensional spectrum. The units of both axes are chemical shifts. The COSY and TOCSY transfer magnetization through the chemical bonds between adjacent protons. The conventional correlation spectroscopy experiment is only able to transfer magnetization between protons on adjacent atoms, whereas in the total correlation spectroscopy experiment the protons are able to relay the magnetization, so it is transferred among all the protons that are connected by adjacent atoms. Thus in a conventional correlation spectroscopy, an alpha proton transfers magnetization to the beta protons, the beta protons transfers to the alpha and gamma protons, if any are present, then the gamma proton transfers to the beta and the delta protons, and the process continues. In total correlation spectroscopy, the alpha and all the other protons are able to transfer magnetization to the beta, gamma, delta, epsilon if they are connected by a continuous chain of protons. The continuous chain of protons are the sidechain of the individual amino acid
s. Thus these two experiments are used to build so called spin systems, that is build a list of resonances of the chemical shift of the peptide proton, the alpha protons and all the protons from each residue
’s sidechain. Which chemical shifts corresponds to which nuclei in the spin system is determined by the conventional correlation spectroscopy connectivities and the fact that different types of protons have characteristic chemical shifts. To connect the different spinsystems in a sequential order, the nuclear Overhauser effect spectroscopy experiment has to be used. Because this experiment transfers magnetization through space, it will show crosspeaks for all protons that are close in space regardless of whether they are in the same spin system or not. The neighbouring residues are inherently close in space, so the assignments can be made by the peaks in the NOESY with other spin systems.
One important problem using homonuclear nuclear magnetic resonance is overlap
between peaks. This occurs when different protons have the same or very similar chemical shifts. This problem becomes greater as the protein becomes larger, so homonuclear nuclear magnetic resonance is usually restricted to small proteins or peptides.

, HN(CO)CA
, HNCACB and CBCA(CO)NH. All six experiments consist of a 1H-15N plane (similar to a HSQC spectrum) expanded with a carbon dimension. In the HN(CA)CO, each HN plane contains the peaks from the carbonyl carbon from its residue as well the preceding one in the sequence. The HNCO contains the carbonyl carbon chemical shift from only the preceding residue, but is much more sensitive than HN(CA)CO. These experiments allow each 1H-15N peak to be linked to the preceding carbonyl carbon, and sequential assignment can then be undertaken by matching the shifts of each spin system's own and previous carbons. The HNCA and HN(CO)CA works similarly, just with the alpha carbons (Cα) rather than the carbonyls, and the HNCACB and the CBCA(CO)NH contains both the alpha carbon and the beta carbon (Cβ). Usually several of these experiments are required to resolve overlap in the carbon dimension. This procedure is usually less ambiguous than the NOESY based method, since it is based on through bond transfer. In the NOESY-based methods additional peaks that are close in space but not belonging to the sequential residues will appear confusing the assignment process. When the sequential assignment has been done it is usually possible to extend the assignment from the Cα and Cβ to the rest of the sidechain using experiments such as HCCH-TOCSY, which is basically a TOCSY experiment resolved in an additional carbon dimension.
It is of great importance to assign the noesy peaks to the correct nuclei based on the chemical shifts. If this task is performed manually it is usually very labor intensive, since proteins usually have thousands of noesy peaks. Some computer programs such as UNIO
, CYANA
and ARIA/CNS perform this task automatically on manually pre-processed listings of peak positions and peak volumes, coupled to a structure calculation. Direct access to the raw NOESY data without the cumbersome need of iteratively refined peak lists is so far only granted by the ATNOS/CANDID approach implemented in the UNIO software package and thus indeed guarantees objective and efficient NOESY spectral analysis.
To obtain as accurate assignments as possible it is a great advantage to have access to carbon-13 and nitrogen-15 noesy experiments, since they help to resolve overlap in the proton dimension. This leads to faster and more reliable assignments, and in turn to better structures.
, to generate angle restraints from coupling constant
s. Another approach uses the chemical shifts to generate angle restraints. Both methods use the fact that the geometry around the alpha carbon affects the coupling constants and chemical shifts, so given the coupling constants or the chemical shifts, a qualified guess can be made about the torsion angles.
 The analyte molecules in a sample can be partially ordered with respect to the external magnetic field of the spectrometer by manipulating the sample conditions. Common techniques include addition of bacteriophage
The analyte molecules in a sample can be partially ordered with respect to the external magnetic field of the spectrometer by manipulating the sample conditions. Common techniques include addition of bacteriophage
s or bicelles to the sample, or preparation of the sample in a stretched polyacrylamide gel. This creates a local environment that favours certain orientations of nonspherical molecules. Normally in solution NMR the dipolar couplings between nuclei are averaged out because of the fast tumbling of the molecule. The slight overpopulation of one orientation means that a residual dipolar coupling
remains to be observed. The dipolar coupling is commonly used in solid state NMR
and provides information about the relative orientation of the bond vectors relative to a single global reference frame. Typically the orientation of the N-H vector is probed in a HSQC like experiment. Initially residual dipolar couplings were used for refinement of previously determined structures, but attempts at de novo structure determination have also been made.
, the reaction can be monitored by NMR spectroscopy. How rapidly a given amide exchanges reflects its solvent accessibility. Thus amide exchange rates can give information on which parts of the protein are buried, hydrogen bonded etc. A common application is to compare the exchange of a free form versus a complex. The amides that become protected in the complex, are assumed to be in the interaction interface.
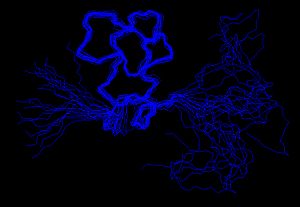 The experimentially determined restraints can be used as input for the structure calculation process. Researchers, using computer programs such as CYANA (Software)
The experimentially determined restraints can be used as input for the structure calculation process. Researchers, using computer programs such as CYANA (Software)
or XPLOR-NIH
, attempt to satisfy as many of the restraints as possible, in addition to general properties of proteins such as bond lengths and angles. The algorithms convert the restraints and the general protein properties into energy terms, and thus tries to minimize the energy. The process results in an ensemble of structures that, if the data were sufficient to dictate a certain fold, will converge.
can yield information on the dynamics of various parts of the protein
. This usually involves measuring relaxation times such as T1 and T2 to determine order parameters, correlation times, and chemical exchange rates. NMR relaxation is a consequence of local fluctuating magnetic fields within a molecule. Local fluctuating magnetic fields are generated by molecular motions. In this way measurements of relaxation times can provide information of motions within a molecule on the atomic level. In NMR studies of protein dynamics the nitrogen-15 isotope is the preferred nucleus to study because its relaxation times are relatively simple to relate to molecular motions This however requires isotope labeling of the protein. The T1 and T2 relaxation times can be measured using various types of HSQC based experiments. The types of motions which can be detected are motions that occur on a time-scale ranging from about 10 picoseconds to about 10 nanoseconds. In addition slower motions, which take place on a time-scale ranging from about 10 microseconds to 100 milliseconds, can also be studied. However, since nitrogen atoms are mainly found in the backbone of a protein, the results mainly reflect the motions of the backbone, which is the most rigid part of a protein molecule. Thus, the results obtained from nitrogen-15 relaxation measurements may not be representative for the whole protein. Therefore techniques utilizing relaxation measurements of carbon-13
and deuterium
have recently been developed, which enables systematic studies of motions of the amino acid side chains in proteins.
of proteins. By using these techniques it has been possible to study proteins in complex with the 900 kDa chaperone GroES-GroEL
.
). The two most time consuming processes involved are the sequence-specific resonance assignment (backbone and side-chain assignment) and the NOE assignment tasks. Several different computer programs have been published that target individual parts of the overall NMR structure determination process in an automated fashion. Most progress has been achieved for the task of automated NOE assignment. So far, only the FLYA and the UNIO approach were proposed to perform the entire protein NMR structure determination process in an automated manner without any human intervention. Efforts have also been made to standardize the structure calculation protocol to make it quicker and more amenable to automation.
Structural biology
Structural biology is a branch of molecular biology, biochemistry, and biophysics concerned with the molecular structure of biological macromolecules, especially proteins and nucleic acids, how they acquire the structures they have, and how alterations in their structures affect their function...
in which NMR spectroscopy
NMR spectroscopy
Nuclear magnetic resonance spectroscopy, most commonly known as NMR spectroscopy, is a research technique that exploits the magnetic properties of certain atomic nuclei to determine physical and chemical properties of atoms or the molecules in which they are contained...
is used to obtain information about the structure and dynamics of proteins. The field was pioneered by Richard R. Ernst
Richard R. Ernst
Richard Robert Ernst is a Swiss physical chemist and Nobel Laureate.Born in Winterthur, Switzerland, Ernst was awarded the Nobel Prize in Chemistry in 1991 for his contributions towards the development of Fourier Transform nuclear magnetic resonance spectroscopy while at Varian Associates, Palo...
and Kurt Wüthrich
Kurt Wüthrich
Kurt Wüthrich is a Swiss chemist and Nobel Chemistry laureate.-Biography:Born in Aarberg, Switzerland, Wüthrich was educated in chemistry, physics, and mathematics at the University of Berne before pursuing his Ph.D. under the direction of Silvio Fallab at the University of Basel, awarded in 1964...
http://www.nature.com/nsmb/journal/v8/n11/full/nsb1101-923.html, among others. Protein NMR techniques are continually being used and improved in both academia
Academia
Academia is the community of students and scholars engaged in higher education and research.-Etymology:The word comes from the akademeia in ancient Greece. Outside the city walls of Athens, the gymnasium was made famous by Plato as a center of learning...
and the biotech industry
Biotechnology
Biotechnology is a field of applied biology that involves the use of living organisms and bioprocesses in engineering, technology, medicine and other fields requiring bioproducts. Biotechnology also utilizes these products for manufacturing purpose...
. Structure determination by NMR spectroscopy usually consists of several following phases, each using a separate set of highly specialized techniques. The sample is prepared, resonances are assigned, restraints are generated and a structure is calculated and validated.
Sample preparation
Protein purification
Protein purification is a series of processes intended to isolate a single type of protein from a complex mixture. Protein purification is vital for the characterization of the function, structure and interactions of the protein of interest. The starting material is usually a biological tissue or...
protein. Usually the sample consist of between 300 and 600 microlitres with a protein concentration in the range 0.1 – 3 millimolar
Mole (unit)
The mole is a unit of measurement used in chemistry to express amounts of a chemical substance, defined as an amount of a substance that contains as many elementary entities as there are atoms in 12 grams of pure carbon-12 , the isotope of carbon with atomic weight 12. This corresponds to a value...
. The source of the protein can be either natural or produced in an expression system
Protein expression
Protein expression is a subcomponent of gene expression. It consists of the stages after DNA has been translated into polypeptide chains, which are ultimately folded into proteins...
using recombinant DNA
Recombinant DNA
Recombinant DNA molecules are DNA sequences that result from the use of laboratory methods to bring together genetic material from multiple sources, creating sequences that would not otherwise be found in biological organisms...
techniques through genetic engineering
Genetic engineering
Genetic engineering, also called genetic modification, is the direct human manipulation of an organism's genome using modern DNA technology. It involves the introduction of foreign DNA or synthetic genes into the organism of interest...
. Recombinantly expressed
Protein expression
Protein expression is a subcomponent of gene expression. It consists of the stages after DNA has been translated into polypeptide chains, which are ultimately folded into proteins...
proteins are usually easier to produce in sufficient quantity, and makes isotopic
Isotope
Isotopes are variants of atoms of a particular chemical element, which have differing numbers of neutrons. Atoms of a particular element by definition must contain the same number of protons but may have a distinct number of neutrons which differs from atom to atom, without changing the designation...
labelling
Isotopic labeling
Isotopic labeling is a technique for tracking the passage of a sample of substance through a system. The substance is 'labeled' by including unusual isotopes in its chemical composition...
possible.
The purified protein is usually dissolved in a buffer solution
Buffer solution
A buffer solution is an aqueous solution consisting of a mixture of a weak acid and its conjugate base or a weak base and its conjugate acid. It has the property that the pH of the solution changes very little when a small amount of strong acid or base is added to it. Buffer solutions are used as a...
and adjusted to the desired solvent conditions. The NMR sample is prepared in a thin walled glass tube
NMR tube
An NMR tube is a thin glass walled tube used to contain samples in nuclear magnetic resonance spectroscopy. Typically NMR tubes come in 5 mm diameters but 10 mm and 3 mm samples are known. It is important that the tubes are uniformly thick and well-balanced to ensure that NMR tube...
.
Data collection
Protein NMR utilizes multidimensional nuclear magnetic resonance experiments to obtain information about the protein. Ideally, each distinct nucleus in the molecule experiences a distinct chemical environment and thus has a distinct chemical shiftChemical shift
In nuclear magnetic resonance spectroscopy, the chemical shift is the resonant frequency of a nucleus relative to a standard. Often the position and number of chemical shifts are diagnostic of the structure of a molecule...
by which it can be recognized. However, in large molecules such as proteins the number of resonances can typically be several thousand and a one-dimensional spectrum inevitably has incidental overlaps. Therefore multidimensional experiments are performed which correlate the frequencies of distinct nuclei. The additional dimensions decrease the chance of overlap and have a larger information content since they correlate signals from nuclei within a specific part of the molecule. Magnetization is transferred into the sample using pulses of electromagnetic (radiofrequency) energy and between nuclei using delays; the process is described with so-called pulse sequence
Pulse sequence
In Fourier Transform NMR spectroscopy and imaging, a pulse sequence describes a series of radio frequency pulses applied to the sample, such that the free induction decay is related to the characteristic frequencies of the wanted signals. After applying a Fourier Transform, the signal can be...
s. Pulse sequences allow the experimenter to investigate and select specific types of connections between nuclei. The array of nuclear magnetic resonance experiments used on proteins fall in two main categories — one where magnetization is transferred through the chemical bonds, and one where the transfer is through space, irrespective of the bonding structure. The first category is used to assign the different chemical shift
Chemical shift
In nuclear magnetic resonance spectroscopy, the chemical shift is the resonant frequency of a nucleus relative to a standard. Often the position and number of chemical shifts are diagnostic of the structure of a molecule...
s to a specific nucleus, and the second is primarily used to generate the distance restraints used in the structure calculation, and in the assignment with unlabelled protein.
Depending on the concentration of the sample, on the magnetic field of the spectrometer, and on the type of experiment, a single multidimensional nuclear magnetic resonance experiment on a protein sample may take hours or even several days to obtain suitable signal-to-noise ratio through signal averaging, and to allow for sufficient evolution of magnetization transfer through the various dimensions of the experiment. Other things being equal, higher-dimensional experiments will take longer than lower-dimensional experiments.
Typically the first experiment to be measured with an isotope-labelled protein is a 2D heteronuclear single quantum correlation
Heteronuclear single quantum correlation
The Heteronuclear Single Quantum Coherence experiment is used frequently in NMR spectroscopy of organic molecules and is of particular significance in the field of protein NMR. It was invented by Geoffrey Bodenhausen and D. J. Ruben in 1980...
(HSQC) spectrum where "heteronuclear" refers to nuclei other than 1H. In theory the heteronuclear single quantum correlation has one peak for each H bound to a heteronucleus. Thus in the 15N-HSQC one signal is expected for each amino acid residue with the exception of proline
Proline
Proline is an α-amino acid, one of the twenty DNA-encoded amino acids. Its codons are CCU, CCC, CCA, and CCG. It is not an essential amino acid, which means that the human body can synthesize it. It is unique among the 20 protein-forming amino acids in that the α-amino group is secondary...
which has no amide-hydrogen due to the cyclic nature of its backbone. Tryptophan
Tryptophan
Tryptophan is one of the 20 standard amino acids, as well as an essential amino acid in the human diet. It is encoded in the standard genetic code as the codon UGG...
and certain other residues with N-containing sidechains also give rise to additional signals. The 15N-HSQC is often referred to as the fingerprint of a protein because each protein has a unique pattern of signal positions. Analysis of the 15N-HSQC allows researchers to evaluate whether the expected number of peaks is present and thus to identify possible problems due to multiple conformations or sample heterogeneity. The relatively quick heteronuclear single quantum correlation experiment helps determine the feasibility of doing subsequent longer, more expensive, and more elaborate experiments. It is not possible to assign peaks to specific atoms from the heteronuclear single quantum correlation alone.
Resonance assignment
In order to analyze the nuclear magnetic resonance data, it is important to get a resonance assignment for the protein. That is to find out which chemical shiftChemical shift
In nuclear magnetic resonance spectroscopy, the chemical shift is the resonant frequency of a nucleus relative to a standard. Often the position and number of chemical shifts are diagnostic of the structure of a molecule...
corresponds to which atom. This is typically achieved by sequential walking
Sequential Walking
Sequential walking is a technique that can be used to solve various 2D NMR spectra. In a 2D experiment, cross peaks must be correlated to the correct nuclei. Using sequential walking, the correct nuclei can be assigned to their crosspeaks...
using information derived from several different types of NMR experiment. The exact procedure depends on whether the protein is isotopically labelled
Isotopic labeling
Isotopic labeling is a technique for tracking the passage of a sample of substance through a system. The substance is 'labeled' by including unusual isotopes in its chemical composition...
or not, since a lot of the assignment experiments depend on carbon-13 and nitrogen-15.
Homonuclear nuclear magnetic resonance
With unlabelled protein the usual procedure is to record a set of two dimensional homonuclear nuclear magnetic resonance experiments through correlation spectroscopyCorrelation spectroscopy
Two-dimensional nuclear magnetic resonance spectroscopy is a set of nuclear magnetic resonance spectroscopy methods which give data plotted in a space defined by two frequency axes rather than one. Types of 2D NMR include correlation spectroscopy , J-spectroscopy, exchange spectroscopy , and...
(COSY), of which several types include conventional correlation spectroscopy, total correlation spectroscopy (TOCSY) and nuclear Overhauser effect
Nuclear Overhauser effect
The Nuclear Overhauser Effect is the transfer of nuclear spin polarization from one nuclear spin population to another via cross-relaxation. It is a common phenomenon observed by nuclear magnetic resonance spectroscopy. The theoretical basis for the NOE was described and experimentally verified...
spectroscopy (NOESY). A two-dimensional nuclear magnetic resonance experiment produces a two-dimensional spectrum. The units of both axes are chemical shifts. The COSY and TOCSY transfer magnetization through the chemical bonds between adjacent protons. The conventional correlation spectroscopy experiment is only able to transfer magnetization between protons on adjacent atoms, whereas in the total correlation spectroscopy experiment the protons are able to relay the magnetization, so it is transferred among all the protons that are connected by adjacent atoms. Thus in a conventional correlation spectroscopy, an alpha proton transfers magnetization to the beta protons, the beta protons transfers to the alpha and gamma protons, if any are present, then the gamma proton transfers to the beta and the delta protons, and the process continues. In total correlation spectroscopy, the alpha and all the other protons are able to transfer magnetization to the beta, gamma, delta, epsilon if they are connected by a continuous chain of protons. The continuous chain of protons are the sidechain of the individual amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
s. Thus these two experiments are used to build so called spin systems, that is build a list of resonances of the chemical shift of the peptide proton, the alpha protons and all the protons from each residue
Residue (chemistry)
In chemistry, residue is the material remaining after a distillation or an evaporation, or to a portion of a larger molecule, such as a methyl group. It may also refer to the undesired byproducts of a reaction....
’s sidechain. Which chemical shifts corresponds to which nuclei in the spin system is determined by the conventional correlation spectroscopy connectivities and the fact that different types of protons have characteristic chemical shifts. To connect the different spinsystems in a sequential order, the nuclear Overhauser effect spectroscopy experiment has to be used. Because this experiment transfers magnetization through space, it will show crosspeaks for all protons that are close in space regardless of whether they are in the same spin system or not. The neighbouring residues are inherently close in space, so the assignments can be made by the peaks in the NOESY with other spin systems.
One important problem using homonuclear nuclear magnetic resonance is overlap
Overlap
Overlap may mean one of:* In music theory, overlap is a synonym for reinterpretation of a chord at the boundary of two musical phrases.* In railway signalling, an Overlap is the length of track beyond a stop signal that is proved to be clear of vehicles in the controls of the previous signal, as a...
between peaks. This occurs when different protons have the same or very similar chemical shifts. This problem becomes greater as the protein becomes larger, so homonuclear nuclear magnetic resonance is usually restricted to small proteins or peptides.
Nitrogen-15 nuclear magnetic resonance
Carbon-13 and nitrogen-15 nuclear magnetic resonance
When the protein is labelled with carbon-13 and nitrogen-15 it is possible to record experiments that transfers magnetisation over the peptide bond, and thus connect different spin systems through bonds. This is usually done using some of the following experiments, HNCO, HN(CA)CO, HNCAHNCA experiment
HNCA is a 3D NMR experiment commonly used in the field of protein NMR. The name derives from the experiment's magnetization transfer pathway: The magnetization of the amide proton of an amino acid residue is transferred to the amide nitrogen, and then to the alpha carbons of both the starting...
, HN(CO)CA
HNCOCA experiment
HNCOCA is a 3D NMR experiment commonly used in the field of protein NMR. The name derives from the experiment's magnetization transfer pathway: The magnetization of the amide proton of an amino acid residue is transferred to the amide nitrogen, and then to the alpha carbon of the previous residue...
, HNCACB and CBCA(CO)NH. All six experiments consist of a 1H-15N plane (similar to a HSQC spectrum) expanded with a carbon dimension. In the HN(CA)CO, each HN plane contains the peaks from the carbonyl carbon from its residue as well the preceding one in the sequence. The HNCO contains the carbonyl carbon chemical shift from only the preceding residue, but is much more sensitive than HN(CA)CO. These experiments allow each 1H-15N peak to be linked to the preceding carbonyl carbon, and sequential assignment can then be undertaken by matching the shifts of each spin system's own and previous carbons. The HNCA and HN(CO)CA works similarly, just with the alpha carbons (Cα) rather than the carbonyls, and the HNCACB and the CBCA(CO)NH contains both the alpha carbon and the beta carbon (Cβ). Usually several of these experiments are required to resolve overlap in the carbon dimension. This procedure is usually less ambiguous than the NOESY based method, since it is based on through bond transfer. In the NOESY-based methods additional peaks that are close in space but not belonging to the sequential residues will appear confusing the assignment process. When the sequential assignment has been done it is usually possible to extend the assignment from the Cα and Cβ to the rest of the sidechain using experiments such as HCCH-TOCSY, which is basically a TOCSY experiment resolved in an additional carbon dimension.
Restraint generation
In order to make structure calculations a number of experimentally determined restraints have to be generated. These fall into different categories, the most widely used is distance restraints and angle restraints.Distance restraints
A crosspeak in a NOESY experiment signifies spatial proximity between the two nuclei in question. Thus each peak can be converted in to a maximum distance between the nuclei, usually between 1,8 and 6 angstroms. The intensity of a noesy peak is proportional to the distance to the minus 6th power, so the distance is determined according to intensity of the peak. The intensity-distance relationship is not exact, so usually a distance range is used.It is of great importance to assign the noesy peaks to the correct nuclei based on the chemical shifts. If this task is performed manually it is usually very labor intensive, since proteins usually have thousands of noesy peaks. Some computer programs such as UNIO
Unio
Unio can refer to:* Unio, a genus in the family Unionidae * UNIO Satu Mare, machine building company from Romania* Utdanningsgruppenes Hovedorganisasjon , a national trade union center in Norway....
, CYANA
Cyana
Cyana is a genus of moths in the Arctiidae family.-Species:* Cyana aroa Bethune-Baker, 1904* Cyana basialba Rothschild, 1913* Cyana bicolor Rothschild, 1913* Cyana brunnea Bethune-Baker, 1904...
and ARIA/CNS perform this task automatically on manually pre-processed listings of peak positions and peak volumes, coupled to a structure calculation. Direct access to the raw NOESY data without the cumbersome need of iteratively refined peak lists is so far only granted by the ATNOS/CANDID approach implemented in the UNIO software package and thus indeed guarantees objective and efficient NOESY spectral analysis.
To obtain as accurate assignments as possible it is a great advantage to have access to carbon-13 and nitrogen-15 noesy experiments, since they help to resolve overlap in the proton dimension. This leads to faster and more reliable assignments, and in turn to better structures.
Angle restraints
In addition to distance restraints, restraints on the torsion angles of the chemical bonds, typically the psi and phi angles can be generated. One approach is to use the Karplus equationKarplus equation
The Karplus equation, named after Martin Karplus, describes the correlation between 3J-coupling constants and dihedral torsion angles in nuclear magnetic resonance spectroscopy:J = A \cos^2 \phi + B \cos\,\phi + C...
, to generate angle restraints from coupling constant
J-coupling
J-coupling is the coupling between two nuclear spins due to the influence of bonding electrons on the magnetic field running between the two nuclei. J-coupling contains information about dihedral angles, which can be estimated using the Karplus equation...
s. Another approach uses the chemical shifts to generate angle restraints. Both methods use the fact that the geometry around the alpha carbon affects the coupling constants and chemical shifts, so given the coupling constants or the chemical shifts, a qualified guess can be made about the torsion angles.
Orientation restraints
Bacteriophage
A bacteriophage is any one of a number of viruses that infect bacteria. They do this by injecting genetic material, which they carry enclosed in an outer protein capsid...
s or bicelles to the sample, or preparation of the sample in a stretched polyacrylamide gel. This creates a local environment that favours certain orientations of nonspherical molecules. Normally in solution NMR the dipolar couplings between nuclei are averaged out because of the fast tumbling of the molecule. The slight overpopulation of one orientation means that a residual dipolar coupling
Residual dipolar coupling
The residual dipolar coupling between two spins in a molecule occurs if the molecules in solution exhibit a partial alignment leading to an incomplete averaging of spatially anisotropic dipolar couplings....
remains to be observed. The dipolar coupling is commonly used in solid state NMR
Solid-state nuclear magnetic resonance
Solid-state NMR spectroscopy is a kind of nuclear magnetic resonance spectroscopy, characterized by the presence of anisotropic interactions.-Introduction:Basic concepts...
and provides information about the relative orientation of the bond vectors relative to a single global reference frame. Typically the orientation of the N-H vector is probed in a HSQC like experiment. Initially residual dipolar couplings were used for refinement of previously determined structures, but attempts at de novo structure determination have also been made.
Hydrogen–deuterium exchange
NMR spectroscopy is nucleus specific. Thus it can distinguish between hydrogen and deuterium. The amide protons in the protein exchange readily with the solvent, and if the solvent contains a different isotope, typically deuteriumDeuterium
Deuterium, also called heavy hydrogen, is one of two stable isotopes of hydrogen. It has a natural abundance in Earth's oceans of about one atom in of hydrogen . Deuterium accounts for approximately 0.0156% of all naturally occurring hydrogen in Earth's oceans, while the most common isotope ...
, the reaction can be monitored by NMR spectroscopy. How rapidly a given amide exchanges reflects its solvent accessibility. Thus amide exchange rates can give information on which parts of the protein are buried, hydrogen bonded etc. A common application is to compare the exchange of a free form versus a complex. The amides that become protected in the complex, are assumed to be in the interaction interface.
Structure calculation
CYANA (Software)
Combined assignment and dynamics algorithm for NMR applicationsCYANA is a program for automated structure calculation of biological macromolecules on the basis of conformational constraints from NMR...
or XPLOR-NIH
XPLOR-NIH
Xplor-NIH is a biomolecular structure determination program which includes an interface to the legacy X-PLOR program. Restraints derived from many different nuclear magnetic resonance and X-ray scattering experiments can be accommodated during structure calculations....
, attempt to satisfy as many of the restraints as possible, in addition to general properties of proteins such as bond lengths and angles. The algorithms convert the restraints and the general protein properties into energy terms, and thus tries to minimize the energy. The process results in an ensemble of structures that, if the data were sufficient to dictate a certain fold, will converge.
Dynamics
In addition to structures, nuclear magnetic resonanceNuclear magnetic resonance
Nuclear magnetic resonance is a physical phenomenon in which magnetic nuclei in a magnetic field absorb and re-emit electromagnetic radiation...
can yield information on the dynamics of various parts of the protein
Protein
Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form, facilitating a biological function. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of...
. This usually involves measuring relaxation times such as T1 and T2 to determine order parameters, correlation times, and chemical exchange rates. NMR relaxation is a consequence of local fluctuating magnetic fields within a molecule. Local fluctuating magnetic fields are generated by molecular motions. In this way measurements of relaxation times can provide information of motions within a molecule on the atomic level. In NMR studies of protein dynamics the nitrogen-15 isotope is the preferred nucleus to study because its relaxation times are relatively simple to relate to molecular motions This however requires isotope labeling of the protein. The T1 and T2 relaxation times can be measured using various types of HSQC based experiments. The types of motions which can be detected are motions that occur on a time-scale ranging from about 10 picoseconds to about 10 nanoseconds. In addition slower motions, which take place on a time-scale ranging from about 10 microseconds to 100 milliseconds, can also be studied. However, since nitrogen atoms are mainly found in the backbone of a protein, the results mainly reflect the motions of the backbone, which is the most rigid part of a protein molecule. Thus, the results obtained from nitrogen-15 relaxation measurements may not be representative for the whole protein. Therefore techniques utilizing relaxation measurements of carbon-13
Carbon-13
Carbon-13 is a natural, stable isotope of carbon and one of the environmental isotopes. It makes up about 1.1% of all natural carbon on Earth.- Detection by mass spectrometry :...
and deuterium
Deuterium
Deuterium, also called heavy hydrogen, is one of two stable isotopes of hydrogen. It has a natural abundance in Earth's oceans of about one atom in of hydrogen . Deuterium accounts for approximately 0.0156% of all naturally occurring hydrogen in Earth's oceans, while the most common isotope ...
have recently been developed, which enables systematic studies of motions of the amino acid side chains in proteins.
NMR spectroscopy on large proteins
Traditionally nuclear magnetic resonance spectroscopy has been limited to relatively small proteins or protein domains. This is in part caused by problems resolving overlapping peaks in larger proteins, but this has been alleviated by the introduction of isotope labelling and multidimensional experiments. Another more serious problem is the fact that in large proteins the magnetization relaxes faster, which means there is less time to detect the signal. This in turn causes the peaks to become broader and weaker, and eventually disappear. Two techniques have been introduced to attenuate the relaxation: transverse relaxation optimized spectroscopy (TROSY) and deuterationDeuterium
Deuterium, also called heavy hydrogen, is one of two stable isotopes of hydrogen. It has a natural abundance in Earth's oceans of about one atom in of hydrogen . Deuterium accounts for approximately 0.0156% of all naturally occurring hydrogen in Earth's oceans, while the most common isotope ...
of proteins. By using these techniques it has been possible to study proteins in complex with the 900 kDa chaperone GroES-GroEL
GroEL
GroEL belongs to the chaperonin family of molecular chaperones, and is found in a large number of bacteria. It is required for the proper folding of many proteins. To function properly, GroEL requires the lid-like cochaperonin protein complex GroES...
.
Automation of the process
Structure determination by NMR has traditionally been a time consuming process, requiring interactive analysis of the data by a highly trained scientist. There has been a considerable interest in automating the process to increase the throughput of structure determination and to make protein NMR accessible to non-experts (See structural genomicsStructural genomics
Structural genomics seeks to describe the 3-dimensional structure of every protein encoded by a given genome. This genome-based approach allows for a high-throughput method of structure determination by a combination of experimental and modeling approaches...
). The two most time consuming processes involved are the sequence-specific resonance assignment (backbone and side-chain assignment) and the NOE assignment tasks. Several different computer programs have been published that target individual parts of the overall NMR structure determination process in an automated fashion. Most progress has been achieved for the task of automated NOE assignment. So far, only the FLYA and the UNIO approach were proposed to perform the entire protein NMR structure determination process in an automated manner without any human intervention. Efforts have also been made to standardize the structure calculation protocol to make it quicker and more amenable to automation.
See also
- Nuclear magnetic resonance spectroscopy of carbohydrates
- X-ray crystallographyX-ray crystallographyX-ray crystallography is a method of determining the arrangement of atoms within a crystal, in which a beam of X-rays strikes a crystal and causes the beam of light to spread into many specific directions. From the angles and intensities of these diffracted beams, a crystallographer can produce a...
- Nuclear magnetic resonanceNuclear magnetic resonanceNuclear magnetic resonance is a physical phenomenon in which magnetic nuclei in a magnetic field absorb and re-emit electromagnetic radiation...
- NMR spectroscopyNMR spectroscopyNuclear magnetic resonance spectroscopy, most commonly known as NMR spectroscopy, is a research technique that exploits the magnetic properties of certain atomic nuclei to determine physical and chemical properties of atoms or the molecules in which they are contained...
- Protein crystallizationProtein crystallizationMost Proteins and many biological macromolecules differ from "small" molecules because the environment in which they function is aqueous. Therefore most biological macromolecules can be prompted to form crystals when the solution in which they are dissolved becomes supersaturated. The manner in...
- Protein dynamics
External links
- NOESY-Based Strategy for Assignments of Backbone and Side Chain Resonances of Large Proteins without Deuteration (a protocol)
- ProSA-web Web service for the recognition of errors in experimentally or theoretically determined protein structures
- Protein NMR Protein NMR experiments