

Secondary structure
Encyclopedia

Biochemistry
Biochemistry, sometimes called biological chemistry, is the study of chemical processes in living organisms, including, but not limited to, living matter. Biochemistry governs all living organisms and living processes...
and structural biology
Structural biology
Structural biology is a branch of molecular biology, biochemistry, and biophysics concerned with the molecular structure of biological macromolecules, especially proteins and nucleic acids, how they acquire the structures they have, and how alterations in their structures affect their function...
, secondary structure is the general three-dimensional form of local segments of biopolymer
Biopolymer
Biopolymers are polymers produced by living organisms. Since they are polymers, Biopolymers contain monomeric units that are covalently bonded to form larger structures. There are three main classes of biopolymers based on the differing monomeric units used and the structure of the biopolymer formed...
s such as protein
Protein
Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form, facilitating a biological function. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of...
s and nucleic acid
Nucleic acid
Nucleic acids are biological molecules essential for life, and include DNA and RNA . Together with proteins, nucleic acids make up the most important macromolecules; each is found in abundance in all living things, where they function in encoding, transmitting and expressing genetic information...
s (DNA/RNA). It does not, however, describe specific atomic positions in three-dimensional space, which are considered to be tertiary structure
Tertiary structure
In biochemistry and molecular biology, the tertiary structure of a protein or any other macromolecule is its three-dimensional structure, as defined by the atomic coordinates.-Relationship to primary structure:...
.
Secondary structure can be formally defined by the hydrogen bond
Hydrogen bond
A hydrogen bond is the attractive interaction of a hydrogen atom with an electronegative atom, such as nitrogen, oxygen or fluorine, that comes from another molecule or chemical group. The hydrogen must be covalently bonded to another electronegative atom to create the bond...
s of the biopolymer, as observed in an atomic-resolution structure. In proteins, the secondary structure is defined by the patterns of hydrogen bonds between backbone amide and carboxyl groups. In nucleic acids, the secondary structure is defined by the hydrogen bonding between the nitrogenous bases. The hydrogen bonding patterns may be significantly distorted, which makes an automatic determination of secondary structure
DSSP (protein)
The DSSP algorithm is the standard method for assigning secondary structure to the amino acids of a protein, given the atomic-resolution coordinates of the protein...
difficult.
The secondary structure may be also defined based on the regular pattern of backbone dihedral angle
Dihedral angle
In geometry, a dihedral or torsion angle is the angle between two planes.The dihedral angle of two planes can be seen by looking at the planes "edge on", i.e., along their line of intersection...
s in a particular region of the Ramachandran plot
Ramachandran plot
-Introduction and early history:A Ramachandran plot , originally developed in 1963 by G. N. Ramachandran C. Ramakrishnan and V...
; thus, a segment of residues with such dihedral angles may be called a helix, regardless of whether it has the correct hydrogen bonds. The secondary structure may be also provided by crystallographers in the corresponding PDB
Protein Data Bank (file format)
The Protein Data Bank file format is a textual file format describing the three dimensional structures of molecules held in the Protein Data Bank. The pdb format accordingly provides for description and annotation of protein and nucleic acid structures including atomic coordinates, observed...
file.
The rough secondary-structure content of a biopolymer (e.g., "this protein is 40% α-helix
Alpha helix
A common motif in the secondary structure of proteins, the alpha helix is a right-handed coiled or spiral conformation, in which every backbone N-H group donates a hydrogen bond to the backbone C=O group of the amino acid four residues earlier...
and 20% β-sheet
Beta sheet
The β sheet is the second form of regular secondary structure in proteins, only somewhat less common than the alpha helix. Beta sheets consist of beta strands connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet...
.")
can often be estimated spectroscopically
Spectroscopy
Spectroscopy is the study of the interaction between matter and radiated energy. Historically, spectroscopy originated through the study of visible light dispersed according to its wavelength, e.g., by a prism. Later the concept was expanded greatly to comprise any interaction with radiative...
. For proteins, a common method is far-ultraviolet
(far-UV, 170-250 nm) circular dichroism
Circular dichroism
Circular dichroism refers to the differential absorption of left and right circularly polarized light. This phenomenon was discovered by Jean-Baptiste Biot, Augustin Fresnel, and Aimé Cotton in the first half of the 19th century. It is exhibited in the absorption bands of optically active chiral...
. A pronounced double minimum at 208 and 222 nm indicate α-helical
structure, whereas a single minimum at 204 nm or 217 nm reflects random-coil or β-sheet structure, respectively.
A less common method is infrared
Infrared
Infrared light is electromagnetic radiation with a wavelength longer than that of visible light, measured from the nominal edge of visible red light at 0.74 micrometres , and extending conventionally to 300 µm...
spectroscopy, which detects differences in the bond
oscillations of amide groups due to hydrogen-bonding. Finally, secondary-structure contents may be
estimated accurately using the chemical shift
Chemical shift
In nuclear magnetic resonance spectroscopy, the chemical shift is the resonant frequency of a nucleus relative to a standard. Often the position and number of chemical shifts are diagnostic of the structure of a molecule...
s of an unassigned NMR
Nuclear magnetic resonance
Nuclear magnetic resonance is a physical phenomenon in which magnetic nuclei in a magnetic field absorb and re-emit electromagnetic radiation...
spectrum.
Secondary structure was introduced by Kaj Ulrik Linderstrøm-Lang at Stanford in 1952.
Protein
Secondary structure in proteins consists of local inter-residue interactions mediated by hydrogen bonds, or not. The most common secondary structures are alpha helicesAlpha helix
A common motif in the secondary structure of proteins, the alpha helix is a right-handed coiled or spiral conformation, in which every backbone N-H group donates a hydrogen bond to the backbone C=O group of the amino acid four residues earlier...
and beta sheet
Beta sheet
The β sheet is the second form of regular secondary structure in proteins, only somewhat less common than the alpha helix. Beta sheets consist of beta strands connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet...
s. Other helices, such as the 310 helix and π helix
Pi helix
A pi helix is a type of secondary structure found in proteins. Although thought to be rare, π-helices are actually found in 15% of known protein structures and are believed to be an evolutionary adaptation derived by the insertion of a single amino acid into an α-helix...
, are calculated to have energetically favorable hydrogen-bonding patterns but are rarely if ever observed in natural proteins except at the ends of α helices due to unfavorable backbone packing in the center of the helix. Other extended structures such as the polyproline helix
Polyproline helix
A Polyproline Helix is a type of protein secondary structure, which occurs in proteins comprising repeating proline residues. A left-handed polyproline II helix is formed when sequential residues all adopt backbone dihedral angles of roughly and have trans isomers of their peptide bonds...
and alpha sheet
Alpha sheet
The alpha sheet is a hypothetical secondary structure in proteins, first proposed by Linus Pauling and Robert Corey in 1951...
are rare in native state
Native state
In biochemistry, the native state of a protein is its operative or functional form. While all protein molecules begin as simple unbranched chains of amino acids, once completed they assume highly specific three-dimensional shapes; that ultimate shape, known as tertiary structure, is the folded...
proteins but are often hypothesized as important protein folding
Protein folding
Protein folding is the process by which a protein structure assumes its functional shape or conformation. It is the physical process by which a polypeptide folds into its characteristic and functional three-dimensional structure from random coil....
intermediates. Tight turns
Turn (biochemistry)
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.- Definition :According to the most common definition, a turn is a structural motif where the Cα atoms of two residues separated by few peptide bonds are in close approach A turn is...
and loose, flexible loops link the more "regular" secondary structure elements. The random coil
Random coil
A random coil is a polymer conformation where the monomer subunits are oriented randomly while still being bonded to adjacent units. It is not one specific shape, but a statistical distribution of shapes for all the chains in a population of macromolecules...
is not a true secondary structure, but is the class of conformations that indicate an absence of regular secondary structure.
Amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
s vary in their ability to form the various secondary structure elements. Proline
Proline
Proline is an α-amino acid, one of the twenty DNA-encoded amino acids. Its codons are CCU, CCC, CCA, and CCG. It is not an essential amino acid, which means that the human body can synthesize it. It is unique among the 20 protein-forming amino acids in that the α-amino group is secondary...
and glycine
Glycine
Glycine is an organic compound with the formula NH2CH2COOH. Having a hydrogen substituent as its 'side chain', glycine is the smallest of the 20 amino acids commonly found in proteins. Its codons are GGU, GGC, GGA, GGG cf. the genetic code.Glycine is a colourless, sweet-tasting crystalline solid...
are sometimes known as "helix breakers" because they disrupt the regularity of the α helical backbone conformation; however, both have unusual conformational abilities and are commonly found in turns
Turn (biochemistry)
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.- Definition :According to the most common definition, a turn is a structural motif where the Cα atoms of two residues separated by few peptide bonds are in close approach A turn is...
. Amino acids that prefer to adopt helical
Alpha helix
A common motif in the secondary structure of proteins, the alpha helix is a right-handed coiled or spiral conformation, in which every backbone N-H group donates a hydrogen bond to the backbone C=O group of the amino acid four residues earlier...
conformations in proteins include methionine
Methionine
Methionine is an α-amino acid with the chemical formula HO2CCHCH2CH2SCH3. This essential amino acid is classified as nonpolar. This amino-acid is coded by the codon AUG, also known as the initiation codon, since it indicates mRNA's coding region where translation into protein...
, alanine
Alanine
Alanine is an α-amino acid with the chemical formula CH3CHCOOH. The L-isomer is one of the 20 amino acids encoded by the genetic code. Its codons are GCU, GCC, GCA, and GCG. It is classified as a nonpolar amino acid...
, leucine
Leucine
Leucine is a branched-chain α-amino acid with the chemical formula HO2CCHCH2CH2. Leucine is classified as a hydrophobic amino acid due to its aliphatic isobutyl side chain. It is encoded by six codons and is a major component of the subunits in ferritin, astacin and other 'buffer' proteins...
, glutamate and lysine
Lysine
Lysine is an α-amino acid with the chemical formula HO2CCH4NH2. It is an essential amino acid, which means that the human body cannot synthesize it. Its codons are AAA and AAG....
("MALEK" in amino-acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
1-letter codes); by contrast, the large aromatic residues (tryptophan
Tryptophan
Tryptophan is one of the 20 standard amino acids, as well as an essential amino acid in the human diet. It is encoded in the standard genetic code as the codon UGG...
, tyrosine
Tyrosine
Tyrosine or 4-hydroxyphenylalanine, is one of the 22 amino acids that are used by cells to synthesize proteins. Its codons are UAC and UAU. It is a non-essential amino acid with a polar side group...
and phenylalanine
Phenylalanine
Phenylalanine is an α-amino acid with the formula C6H5CH2CHCOOH. This essential amino acid is classified as nonpolar because of the hydrophobic nature of the benzyl side chain. L-Phenylalanine is an electrically neutral amino acid, one of the twenty common amino acids used to biochemically form...
) and
 -branched amino acids (isoleucine
-branched amino acids (isoleucineIsoleucine
Isoleucine is an α-amino acid with the chemical formula HO2CCHCHCH2CH3. It is an essential amino acid, which means that humans cannot synthesize it, so it must be ingested. Its codons are AUU, AUC and AUA....
, valine
Valine
Valine is an α-amino acid with the chemical formula HO2CCHCH2. L-Valine is one of 20 proteinogenic amino acids. Its codons are GUU, GUC, GUA, and GUG. This essential amino acid is classified as nonpolar...
, and threonine
Threonine
Threonine is an α-amino acid with the chemical formula HO2CCHCHCH3. Its codons are ACU, ACA, ACC, and ACG. This essential amino acid is classified as polar...
) prefer to adopt β-strand
Beta sheet
The β sheet is the second form of regular secondary structure in proteins, only somewhat less common than the alpha helix. Beta sheets consist of beta strands connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet...
conformations. However, these preferences are not strong enough to produce a reliable method of predicting secondary structure from sequence alone.
There are several methods for defining protein secondary structure (e.g. DEFINE, DSSP, STRIDE (protein)
STRIDE (protein)
In protein structure, STRIDE is an algorithm for the assignment of protein secondary structure elements given the atomic coordinates of the protein, as defined by X-ray crystallography, protein NMR, or another protein structure determination method...
).
| Geometry attribute | α-helix | 310 helix | π-helix |
|---|---|---|---|
| Residues per turn | 3.6 | 3.0 | 4.4 |
| Translation per residue | 1.5 Å (0.15 nm) | 2 Å (0.2 nm) | 1.1 Å (0.11 nm) |
| Radius of helix | 2.3 Å (0.23 nm) | 1.9 Å (0.19 nm) | 2.8 Å (0.28 nm) |
| Pitch | 5.4 Å (0.54 nm) | 6 Å (0.6 nm) | 4.8 Å (0.48 nm) |
The DSSP code
The Dictionary of Protein Secondary Structure, in short DSSP, is commonly used to describe the protein secondary structure with single letter codes. The secondary structure is assigned based on hydrogen bonding patterns as those initially proposed by Pauling et al. in 1951 (before any protein structureProtein structure
Proteins are an important class of biological macromolecules present in all organisms. Proteins are polymers of amino acids. Classified by their physical size, proteins are nanoparticles . Each protein polymer – also known as a polypeptide – consists of a sequence formed from 20 possible L-α-amino...
had ever been experimentally determined). There are eight types of secondary structure that DSSP defines:
- G = 3-turn helix (310 helix3 10 helixA 310 helix is a type of secondary structure found in proteins.-Structure:The amino acids in a 310-helix are arranged in a right-handed helical structure...
). Min length 3 residues. - H = 4-turn helix (α helix). Min length 4 residues.
- I = 5-turn helix (π helix). Min length 5 residues.
- T = hydrogen bonded turn (3, 4 or 5 turn)
- E = extended strand in parallel and/or anti-parallel β-sheet conformation. Min length 2 residues.
- B = residue in isolated β-bridge (single pair β-sheet hydrogen bond formation)
- S = bend (the only non-hydrogen-bond based assignment).
Amino acid residues which are not in any of the above conformations are assigned as the eighth type 'Coil': often codified as ' ' (space), C (coil) or '-' (dash). The helices (G,H and I) and sheet conformations are all required to have a reasonable length. This means that 2 adjacent residues in the primary structure must form the same hydrogen bonding pattern. If the helix or sheet hydrogen bonding pattern is too short they are designated as T or B, respectively. Other protein secondary structure assignment categories exist (sharp turns, Omega loops etc.), but they are less frequently used.
DSSP H-bond definition
Secondary structure is defined by hydrogen bondHydrogen bond
A hydrogen bond is the attractive interaction of a hydrogen atom with an electronegative atom, such as nitrogen, oxygen or fluorine, that comes from another molecule or chemical group. The hydrogen must be covalently bonded to another electronegative atom to create the bond...
ing, so the exact definition of a hydrogen bond is critical. The standard H-bond definition for secondary structure is that of DSSP
DSSP
DSSP may refer to:*Dessert spoon, a spoon with a capacity of about 2 teaspoons*DSSP , a programming language*DSSP , a method of scanning objects into 3D digital representations...
, which is a purely electrostatic model. It assigns charges of
 to the carbonyl carbon and oxygen, respectively, and charges of
to the carbonyl carbon and oxygen, respectively, and charges of  to the amide nitrogen and hydrogen, respectively. The electrostatic energy is
to the amide nitrogen and hydrogen, respectively. The electrostatic energy is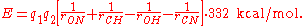
According to DSSP
DSSP
DSSP may refer to:*Dessert spoon, a spoon with a capacity of about 2 teaspoons*DSSP , a programming language*DSSP , a method of scanning objects into 3D digital representations...
, an H-bond exists if and only if
 is less than -0.5 kcal/mol. Although the DSSP formula is a relatively crude approximation of the physical H-bond energy, it is generally accepted as a tool for defining secondary structure.
is less than -0.5 kcal/mol. Although the DSSP formula is a relatively crude approximation of the physical H-bond energy, it is generally accepted as a tool for defining secondary structure.Protein secondary-structure prediction
Predicting protein tertiary structure from only its amino acid sequence is a very challenging problem (see protein structure predictionProtein structure prediction
Protein structure prediction is the prediction of the three-dimensional structure of a protein from its amino acid sequence — that is, the prediction of its secondary, tertiary, and quaternary structure from its primary structure. Structure prediction is fundamentally different from the inverse...
), but using the simpler secondary structure definitions is more tractable and has been the focus for research for a long time.
Although, the 8-state DSSP code is already a simplification from the continuous variation of hydrogen bonding patterns present in a protein the majority of secondary prediction methods simplify further to the three dominant states: Helix, Sheet and Coil. How the conversion is made from 8- to 3-state varies between methods. Early methods of secondary-structure prediction were based on the helix- or sheet-forming propensities of individual amino acids, sometimes coupled with rules for estimating the free energy of forming secondary structure elements. Such methods were typically ~60% accurate in predicting which of the three states (helix/sheet/coil) a residue adopts. A significant increase in accuracy (to nearly ~80%) was made by exploiting multiple sequence alignment
Multiple sequence alignment
A multiple sequence alignment is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a lineage and are descended from a common ancestor...
; knowing the full distribution of amino acids that occur at a position (and in its vicinity, typically ~7 residues on either side) throughout evolution
Evolution
Evolution is any change across successive generations in the heritable characteristics of biological populations. Evolutionary processes give rise to diversity at every level of biological organisation, including species, individual organisms and molecules such as DNA and proteins.Life on Earth...
provides a much better picture of the structural tendencies near that position. For illustration, a given protein might have a glycine
Glycine
Glycine is an organic compound with the formula NH2CH2COOH. Having a hydrogen substituent as its 'side chain', glycine is the smallest of the 20 amino acids commonly found in proteins. Its codons are GGU, GGC, GGA, GGG cf. the genetic code.Glycine is a colourless, sweet-tasting crystalline solid...
at a given position, which by itself might suggest a random coil there. However, multiple sequence alignment might reveal that helix-favoring amino acids occur at that position (and nearby positions) in 95% of homologous proteins spanning nearly a billion years of evolution. Moreover, by examining the average hydrophobicity at that and nearby positions, the same alignment might also suggest a pattern of residue solvent accessibility
Accessible surface area
The accessible surface area is the surface area of a biomolecule that is accessible to a solvent. The ASA is usually quoted in square ångstrom . ASA was first described by Lee & Richards in 1971 and is sometimes called the Lee-Richards molecular surface...
consistent with an α-helix. Taken together, these factors would suggest that the glycine of the original protein adopts α-helical structure, rather than random coil. Several types of methods are used to combine all the available data to form a 3-state prediction, including neural network
Neural network
The term neural network was traditionally used to refer to a network or circuit of biological neurons. The modern usage of the term often refers to artificial neural networks, which are composed of artificial neurons or nodes...
s, hidden Markov model
Hidden Markov model
A hidden Markov model is a statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved states. An HMM can be considered as the simplest dynamic Bayesian network. The mathematics behind the HMM was developed by L. E...
s and support vector machine
Support vector machine
A support vector machine is a concept in statistics and computer science for a set of related supervised learning methods that analyze data and recognize patterns, used for classification and regression analysis...
s. Modern prediction methods also provide a confidence score for their predictions at every position.
Secondary-structure prediction methods are continuously benchmarked, e.g., in the EVA experiment. Based on ~270 weeks of testing, the most accurate methods at present are PSIPRED, SAM, PORTER, PROF and SABLE. Interestingly, it does not seem to be possible to improve upon these methods by taking a consensus of them . The chief area for improvement appears to be the prediction of β-strands; residues confidently predicted as β-strand are likely to be so, but the methods are apt to overlook some β-strand segments (false negatives). There is likely an upper limit of ~90% prediction accuracy overall, due to the idiosyncrasies of the standard method (DSSP
DSSP
DSSP may refer to:*Dessert spoon, a spoon with a capacity of about 2 teaspoons*DSSP , a programming language*DSSP , a method of scanning objects into 3D digital representations...
) for assigning secondary-structure classes (helix/strand/coil) to PDB structures, against which the predictions are benchmarked.
Accurate secondary-structure prediction is a key element in the prediction of tertiary structure
Tertiary structure
In biochemistry and molecular biology, the tertiary structure of a protein or any other macromolecule is its three-dimensional structure, as defined by the atomic coordinates.-Relationship to primary structure:...
, in all but the simplest (homology modeling
Protein structure prediction
Protein structure prediction is the prediction of the three-dimensional structure of a protein from its amino acid sequence — that is, the prediction of its secondary, tertiary, and quaternary structure from its primary structure. Structure prediction is fundamentally different from the inverse...
) cases. For example, a confidently predicted pattern of six secondary structure elements βαββαβ is the signature of a ferredoxin
Ferredoxin
Ferredoxins are iron-sulfur proteins that mediate electron transfer in a range of metabolic reactions. The term "ferredoxin" was coined by D.C. Wharton of the DuPont Co...
fold.
Alignment
Both protein and nucleic acid secondary structures can be used to aid in multiple sequence alignmentMultiple sequence alignment
A multiple sequence alignment is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a lineage and are descended from a common ancestor...
. These alignments can be made more accurate by the inclusion of secondary structure information in addition to simple sequence information. This is sometimes less useful in RNA because base pairing is much more highly conserved than sequence. Distant relationships between proteins whose primary structures are unalignable can sometimes be found by secondary structure.
See also
- Folding (chemistry)Folding (chemistry)In chemistry, folding is the process by which a molecule assumes its shape or conformation. The process can also be described as intramolecular self-assembly where the molecule is directed to form a specific shape through noncovalent interactions, such as hydrogen bonding, metal coordination,...
- protein primary structure
- protein tertiary structure
- protein quaternary structure
- translation
- structural motifStructural motifIn a chain-like biological molecule, such as a protein or nucleic acid, a structural motif is a supersecondary structure, which appears also in a variety of other molecules...
Further reading
- C Branden and J Tooze (1999). Introduction to Protein Structure 2nd ed. Garland Publishing: New York, NY.
- M. Zuker "Computer prediction of RNA structure", Methods in Enzymology, 180:262-88 (1989). (The classic paper on dynamic programming algorithms to predict RNA secondary structure.)
- L. Pauling and R.B Corey. Configurations of polypeptide chains with favored orientations of the polypeptide around single bonds: Two pleated sheets. Proc. Natl. Acad. Sci. Wash., 37:729-740 (1951). (The original beta-sheet conformation article.)
- L. Pauling, R.B. Corey and H.R. Branson. Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. Wash., 37:205-211 (1951). (alpha- and pi-helix conformations, since they predicted that
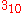 helices would not be possible.)
helices would not be possible.)
External links
- NetSurfP - Secondary Structure and Surface Accessibility predictor
- PROF
- Jpred
- PSIPRED
- DSSP
- WhatIf
- Mfold
- STRIDESTRIDE (protein)In protein structure, STRIDE is an algorithm for the assignment of protein secondary structure elements given the atomic coordinates of the protein, as defined by X-ray crystallography, protein NMR, or another protein structure determination method...
- PSSpred A multiple neural network training program for protein secondary strucure prediction