

DNA sequencing theory
Encyclopedia
DNA sequencing theory is the broad body of work that attempts to lay analytical foundations for DNA sequencing
. The practical aspects revolve around designing and optimizing sequencing projects (known as "strategic genomics"), predicting project performance, troubleshooting experimental results, characterizing factors such as sequence bias and the effects of software processing algorithms, and comparing various sequencing methods to one another. In this sense, it could be considered a branch of systems engineering
or operations research
. The permanent archive of work is primarily mathematical, although numerical calculations are often conducted for particular problems too. DNA sequencing theory addresses physical processes related to sequencing DNA and should not be confused with theories of analyzing resultant DNA sequences, e.g. sequence alignment
. Publications sometimes do not make a careful distinction, but the latter are primarily concerned with algorithm
ic issues.
rely on reading small fragments of DNA and subsequently reconstructing these data to infer the original DNA target, either via assembly
or alignment
to a reference. The abstraction
common to these methods is that of a mathematical covering problem
. For example, one can imagine a line segment representing the target and a subsequent process where smaller segments are "dropped" onto random locations of the target. The target is considered "sequenced" when adequate coverage accumulates, for example when no gaps remain.
The abstract properties of covering have been studied by mathematicians for over a century. However, direct application of these results has not generally been possible. Closed-form mathematical solutions, especially for probability distributions, often cannot be readily evaluated. That is, they involve inordinately large amounts of computer time for parameters characteristic of DNA sequencing
. Stevens' configuration is one such example. Results obtained from the perspective of pure mathematics
also do not account for factors that are actually important in sequencing, for instance detectable overlap in sequencing fragments, double-stranding, edge-effects, and target multiplicity. Consequently, development of sequencing theory has proceeded more according to the philosophy of applied mathematics
. In particular, it has been problem-focused and makes expedient use of approximations, simulations, etc.
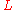 and
and  as the fragment length and target length, respectively, then the probability of "covering" any given location on the target with one particular fragment is
as the fragment length and target length, respectively, then the probability of "covering" any given location on the target with one particular fragment is 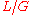 . Note that this presumes
. Note that this presumes  , which is valid for many, though not all sequencing scenarios. Utilizing concepts from the binomial distribution, it can then be shown that the probability that the location is covered by at least one of
, which is valid for many, though not all sequencing scenarios. Utilizing concepts from the binomial distribution, it can then be shown that the probability that the location is covered by at least one of  fragments is
fragments is
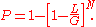
This equation was first used to characterize plasmid libraries, but is often more useful in a modified form. For most projects , so that, to a good degree of approximation
, so that, to a good degree of approximation

where is called the redundancy. Note the significance of redundancy as representing the average number of times a position is covered with fragments. Note also that in considering the covering process over all positions in the target, this probability is identical to the expected value
is called the redundancy. Note the significance of redundancy as representing the average number of times a position is covered with fragments. Note also that in considering the covering process over all positions in the target, this probability is identical to the expected value
of the random variable , which represents the fraction of the target coverage. The final result,
, which represents the fraction of the target coverage. The final result,

remains in widespread use as a "back of the envelope
" estimator and predicts that coverage for all projects evolves along a universal curve that is a function only of the redundancy.
and Michael Waterman
published an important paper examining the covering problem from the standpoint of gaps. Although they focused on the so-called mapping problem
, the abstraction to sequencing is much the same. They furnished a number of useful results that were adopted as the standard theory from the earliest days of "large-scale" genome sequencing. Their model was also used in designing the Human Genome Project
and continues to play an important role in DNA sequencing.
Ultimately, the main goal of a sequencing project is to close all gaps, so the "gap perspective" was a logical basis of developing a sequencing model. One of the more frequently used results from this model is the expected number of contig
s, given the number of fragments sequenced. If one neglects the amount of sequence that is essentially "wasted" by having to detect overlaps, their theory yields
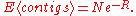
In 1995, Roach published improvements to this theory, enabling it to be applied to sequencing projects in which the goal was to completely sequence a target genome. Wendl and Waterston confirmed, based on Stevens' method, that both models produced similar results when the number of contigs was substantial, such as in low coverage mapping or sequencing projects. As sequencing projects ramped up in the 1990s, and projects approached completion, low coverage approximations became inadequate, and the exact model of Roach was necessary. However, as the cost of sequencing dropped, parameters of sequencing projects became easier to directly test empirically, and interest and funding for strategic genomics diminished
The basic ideas of Lander-Waterman theory led to a number of additional results for particular variations in mapping techniques. However, technological advancements have rendered mapping theories largely obsolete except in organisms other than highly studied model organisms (e.g., yeast, flies, mice, and humans).
and medical (cancer) sequencing
. There are important factors in these scenarios that classical theory does not account for. Recent work has begun to focus on resolving the effects of some of these issues. The level of mathematics becomes commensurately more sophisticated.
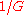 chance of being covered, as opposed to
chance of being covered, as opposed to 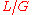 for interior positions. For
for interior positions. For  , classical Lander-Waterman Theory still gives good predictions, but dynamics change for higher redundancies.
, classical Lander-Waterman Theory still gives good predictions, but dynamics change for higher redundancies.
is important and this can only be done if the sequence of the diploid genome is obtained. In the pioneering efforts to sequence individuals, Levy et al. and Wheeler et al., who sequenced Craig Venter
and Jim Watson
, respectively, outlined models for covering both alleles in a genome. Wendl and Wilson followed with a more general theory that allowed for an arbitrary number of coverings of each allele and arbitrary ploidy
. These results point to the general conclusion that the amount of data needed for such projects is significantly higher than for traditional haploid projects.
, biology
, and systems engineering
, so it is highly interdisciplinary. Although many universities now have programs in computational biology
, there does not yet seem to be a strong focus at the graduate level on this topic. Academic contributions have mainly been limited to a small number of PhD dissertations.
DNA sequencing
DNA sequencing includes several methods and technologies that are used for determining the order of the nucleotide bases—adenine, guanine, cytosine, and thymine—in a molecule of DNA....
. The practical aspects revolve around designing and optimizing sequencing projects (known as "strategic genomics"), predicting project performance, troubleshooting experimental results, characterizing factors such as sequence bias and the effects of software processing algorithms, and comparing various sequencing methods to one another. In this sense, it could be considered a branch of systems engineering
Systems engineering
Systems engineering is an interdisciplinary field of engineering that focuses on how complex engineering projects should be designed and managed over the life cycle of the project. Issues such as logistics, the coordination of different teams, and automatic control of machinery become more...
or operations research
Operations research
Operations research is an interdisciplinary mathematical science that focuses on the effective use of technology by organizations...
. The permanent archive of work is primarily mathematical, although numerical calculations are often conducted for particular problems too. DNA sequencing theory addresses physical processes related to sequencing DNA and should not be confused with theories of analyzing resultant DNA sequences, e.g. sequence alignment
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
. Publications sometimes do not make a careful distinction, but the latter are primarily concerned with algorithm
Algorithm
In mathematics and computer science, an algorithm is an effective method expressed as a finite list of well-defined instructions for calculating a function. Algorithms are used for calculation, data processing, and automated reasoning...
ic issues.
Sequencing as a covering problem
All mainstream methods of DNA sequencingDNA sequencing
DNA sequencing includes several methods and technologies that are used for determining the order of the nucleotide bases—adenine, guanine, cytosine, and thymine—in a molecule of DNA....
rely on reading small fragments of DNA and subsequently reconstructing these data to infer the original DNA target, either via assembly
Sequence assembly
In bioinformatics, sequence assembly refers to aligning and merging fragments of a much longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology cannot read whole genomes in one go, but rather reads small pieces of between 20 and 1000 bases,...
or alignment
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
to a reference. The abstraction
Abstraction
Abstraction is a process by which higher concepts are derived from the usage and classification of literal concepts, first principles, or other methods....
common to these methods is that of a mathematical covering problem
Cover (topology)
In mathematics, a cover of a set X is a collection of sets whose union contains X as a subset. Formally, ifC = \lbrace U_\alpha: \alpha \in A\rbrace...
. For example, one can imagine a line segment representing the target and a subsequent process where smaller segments are "dropped" onto random locations of the target. The target is considered "sequenced" when adequate coverage accumulates, for example when no gaps remain.
The abstract properties of covering have been studied by mathematicians for over a century. However, direct application of these results has not generally been possible. Closed-form mathematical solutions, especially for probability distributions, often cannot be readily evaluated. That is, they involve inordinately large amounts of computer time for parameters characteristic of DNA sequencing
DNA sequencing
DNA sequencing includes several methods and technologies that are used for determining the order of the nucleotide bases—adenine, guanine, cytosine, and thymine—in a molecule of DNA....
. Stevens' configuration is one such example. Results obtained from the perspective of pure mathematics
Pure mathematics
Broadly speaking, pure mathematics is mathematics which studies entirely abstract concepts. From the eighteenth century onwards, this was a recognized category of mathematical activity, sometimes characterized as speculative mathematics, and at variance with the trend towards meeting the needs of...
also do not account for factors that are actually important in sequencing, for instance detectable overlap in sequencing fragments, double-stranding, edge-effects, and target multiplicity. Consequently, development of sequencing theory has proceeded more according to the philosophy of applied mathematics
Applied mathematics
Applied mathematics is a branch of mathematics that concerns itself with mathematical methods that are typically used in science, engineering, business, and industry. Thus, "applied mathematics" is a mathematical science with specialized knowledge...
. In particular, it has been problem-focused and makes expedient use of approximations, simulations, etc.
Early uses derived from elementary probability theory
The earliest result was actually borrowed directly from elementary probability theory. If we model the above process and take and as the fragment length and target length, respectively, then the probability of "covering" any given location on the target with one particular fragment is . Note that this presumes , which is valid for many, though not all sequencing scenarios. Utilizing concepts from the binomial distribution, it can then be shown that the probability that the location is covered by at least one of fragments isThis equation was first used to characterize plasmid libraries, but is often more useful in a modified form. For most projects
, so that, to a good degree of approximationwhere
is called the redundancy. Note the significance of redundancy as representing the average number of times a position is covered with fragments. Note also that in considering the covering process over all positions in the target, this probability is identical to the expected valueExpected value
In probability theory, the expected value of a random variable is the weighted average of all possible values that this random variable can take on...
of the random variable
, which represents the fraction of the target coverage. The final result,remains in widespread use as a "back of the envelope
Back-of-the-envelope calculation
A back-of-the-envelope calculation is a rough calculation, typically jotted down on any available scrap of paper such as the actual back of an envelope...
" estimator and predicts that coverage for all projects evolves along a universal curve that is a function only of the redundancy.
Lander-Waterman theory
In 1988, Eric LanderEric Lander
Eric Steven Lander is a Professor of Biology at the Massachusetts Institute of Technology , a member of the Whitehead Institute, and director of the Broad Institute of MIT and Harvard who has devoted his career toward realizing the promise of the human genome for medicine. He is co-chair of U.S...
and Michael Waterman
Michael Waterman
Professor Michael S. Waterman is a scientist at the University of Southern California , where he holds an Endowed Associates Chair in Biological Sciences, Mathematics and Computer Science. He previously held positions at Los Alamos National Laboratory and Idaho State University...
published an important paper examining the covering problem from the standpoint of gaps. Although they focused on the so-called mapping problem
Gene mapping
Gene mapping, also called genome mapping, is the creation of a genetic map assigning DNA fragments to chromosomes.When a genome is first investigated, this map is nonexistent. The map improves with the scientific progress and is perfect when the genomic DNA sequencing of the species has been...
, the abstraction to sequencing is much the same. They furnished a number of useful results that were adopted as the standard theory from the earliest days of "large-scale" genome sequencing. Their model was also used in designing the Human Genome Project
Human Genome Project
The Human Genome Project is an international scientific research project with a primary goal of determining the sequence of chemical base pairs which make up DNA, and of identifying and mapping the approximately 20,000–25,000 genes of the human genome from both a physical and functional...
and continues to play an important role in DNA sequencing.
Ultimately, the main goal of a sequencing project is to close all gaps, so the "gap perspective" was a logical basis of developing a sequencing model. One of the more frequently used results from this model is the expected number of contig
Contig
A contig is a set of overlapping DNA segments that together represent a consensus region of DNA. In bottom-up sequencing projects, a contig refers to overlapping sequence data ; in top-down sequencing projects, contig refers to the overlapping clones that form a physical map of the genome that is...
s, given the number of fragments sequenced. If one neglects the amount of sequence that is essentially "wasted" by having to detect overlaps, their theory yields
In 1995, Roach published improvements to this theory, enabling it to be applied to sequencing projects in which the goal was to completely sequence a target genome. Wendl and Waterston confirmed, based on Stevens' method, that both models produced similar results when the number of contigs was substantial, such as in low coverage mapping or sequencing projects. As sequencing projects ramped up in the 1990s, and projects approached completion, low coverage approximations became inadequate, and the exact model of Roach was necessary. However, as the cost of sequencing dropped, parameters of sequencing projects became easier to directly test empirically, and interest and funding for strategic genomics diminished
The basic ideas of Lander-Waterman theory led to a number of additional results for particular variations in mapping techniques. However, technological advancements have rendered mapping theories largely obsolete except in organisms other than highly studied model organisms (e.g., yeast, flies, mice, and humans).
Parking strategy
The parking strategy for sequencing resembles the process of parking cars along a curb. Each car is a sequenced clone, and the curb is the genomic target. Each clone sequenced is screened to ensure that subsequently sequenced clones do not overlap any previously sequenced clone. No sequencing effort is redundant in this strategy. However, much like the gaps between parked cars, unsequenced gaps less than the length of a clone accumulate between sequenced clones. There can be considerable cost to close such gaps.Pairwise End-sequencing
In 1995, Roach et al. proposed and demonstrated through simulations a generalization of a set of strategies explored earlier by Edwards and Caskey. This whole-genome sequencing method became immensely popular as it was championed by Celera and used to sequenced several model organisms before Celera applied it to the human genome. Today, most sequencing projects employ this strategy, often called paired end sequencing.Recent advancements
The physical processes and protocols of DNA sequencing have continued to evolve, largely driven by advancements in bio-chemical methods, hardware, and automation techniques. There is now a wide range of problems that DNA sequencing has made in-roads into, including metagenomicsMetagenomics
Metagenomics is the study of metagenomes, genetic material recovered directly from environmental samples. The broad field may also be referred to as environmental genomics, ecogenomics or community genomics. Traditional microbiology and microbial genome sequencing rely upon cultivated clonal cultures...
and medical (cancer) sequencing
Cancer Genome Project
The Cancer Genome Project, based at the Wellcome Trust Sanger Institute, aims to identify sequence variants/mutations critical in the development of human cancers...
. There are important factors in these scenarios that classical theory does not account for. Recent work has begun to focus on resolving the effects of some of these issues. The level of mathematics becomes commensurately more sophisticated.
Multiplicity
Biologists have developed methods to filter highly-repetitive, essentially un-sequenceable regions of genomes. These procedures are important for organisms whose genomes consist mostly of such DNA, for example corn. They yield multitudes of small islands of sequenceable DNA products. Wendl and Barbazuk proposed an extension to Lander-Waterman Theory to account for "gaps" in the target due to filtering and the so-called "edge-effect". The latter is a position-specific sampling bias, for example the terminal base position has only a chance of being covered, as opposed to for interior positions. For , classical Lander-Waterman Theory still gives good predictions, but dynamics change for higher redundancies.Small versus large fragments
Modern sequencing methods usually sequence both ends of a larger fragment, which provides linking information for de novo assembly and improved probabilities for alignment to reference sequence. Researchers generally believe that longer lengths of data (read lengths) enhance performance for very large DNA targets, an idea consistent with predictions from distribution models. However, Wendl showed that smaller fragments provide better coverage on small, linear targets because they reduce the edge effect in linear molecules. These findings have implications for sequencing the products of DNA filtering procedures. Read-pairing and fragment size evidently have negligible influence for large, whole-genome class targets.Diploid sequencing
Sequencing is emerging as an important tool in medicine, for example in cancer research. Here, the ability to detect heterozygous mutationsLoss of heterozygosity
Loss of heterozygosity in a cell is the loss of normal function of one allele of a gene in which the other allele was already inactivated. This term is mostly used in the context of oncogenesis; after an inactivating mutation in one allele of a tumor suppressor gene occurs in the parent's germline...
is important and this can only be done if the sequence of the diploid genome is obtained. In the pioneering efforts to sequence individuals, Levy et al. and Wheeler et al., who sequenced Craig Venter
Craig Venter
John Craig Venter is an American biologist and entrepreneur, most famous for his role in being one of the first to sequence the human genome and for his role in creating the first cell with a synthetic genome in 2010. Venter founded Celera Genomics, The Institute for Genomic Research and the J...
and Jim Watson
James D. Watson
James Dewey Watson is an American molecular biologist, geneticist, and zoologist, best known as one of the co-discoverers of the structure of DNA in 1953 with Francis Crick...
, respectively, outlined models for covering both alleles in a genome. Wendl and Wilson followed with a more general theory that allowed for an arbitrary number of coverings of each allele and arbitrary ploidy
Ploidy
Ploidy is the number of sets of chromosomes in a biological cell.Human sex cells have one complete set of chromosomes from the male or female parent. Sex cells, also called gametes, combine to produce somatic cells. Somatic cells, therefore, have twice as many chromosomes. The haploid number is...
. These results point to the general conclusion that the amount of data needed for such projects is significantly higher than for traditional haploid projects.
Limitations
DNA sequencing theories often invoke the assumption that certain random variables in a model are independently and identically distributed. For example, in Lander-Waterman Theory, a sequenced fragment is presumed to have the same probability of covering each region of a genome and all fragments are assumed to be independent of one another. In actuality, sequencing projects are subject to various types of bias, including differences of how well regions can be cloned, sequencing anomalies, biases in the target sequence (which is not random), and software-dependent errors and biases. In general, theory will agree well with observation up to the point that enough data have been generated to expose latent biases. The kinds of biases related to the underlying target sequence are particularly difficult to model, since the sequence itself may not be known a priori. This presents a type of "chicken and egg" closure problem.Academic status
Sequencing theory is based on elements of mathematicsMathematics
Mathematics is the study of quantity, space, structure, and change. Mathematicians seek out patterns and formulate new conjectures. Mathematicians resolve the truth or falsity of conjectures by mathematical proofs, which are arguments sufficient to convince other mathematicians of their validity...
, biology
Biology
Biology is a natural science concerned with the study of life and living organisms, including their structure, function, growth, origin, evolution, distribution, and taxonomy. Biology is a vast subject containing many subdivisions, topics, and disciplines...
, and systems engineering
Systems engineering
Systems engineering is an interdisciplinary field of engineering that focuses on how complex engineering projects should be designed and managed over the life cycle of the project. Issues such as logistics, the coordination of different teams, and automatic control of machinery become more...
, so it is highly interdisciplinary. Although many universities now have programs in computational biology
Computational biology
Computational biology involves the development and application of data-analytical and theoretical methods, mathematical modeling and computational simulation techniques to the study of biological, behavioral, and social systems...
, there does not yet seem to be a strong focus at the graduate level on this topic. Academic contributions have mainly been limited to a small number of PhD dissertations.
See also
- Computational biologyComputational biologyComputational biology involves the development and application of data-analytical and theoretical methods, mathematical modeling and computational simulation techniques to the study of biological, behavioral, and social systems...
- BioinformaticsBioinformaticsBioinformatics is the application of computer science and information technology to the field of biology and medicine. Bioinformatics deals with algorithms, databases and information systems, web technologies, artificial intelligence and soft computing, information and computation theory, software...
- Mathematical biologyMathematical biologyMathematical and theoretical biology is an interdisciplinary scientific research field with a range of applications in biology, medicine and biotechnology...
- Sulston scoreSulston scoreThe Sulston Score is an equation used in DNA mapping to numerically assess the likelihood that a given "fingerprint" similarity between two DNA clones is merely a result of chance. Used as such, it is a test of statistical significance...