

Drug design
Encyclopedia
Drug design, also sometimes referred to as rational
drug design or structure-based drug design, is the inventive
process of finding new medications based on the knowledge of the biological target
. The drug is most commonly an organic
small molecule
that activates or inhibits the function of a biomolecule
such as a protein
, which in turn results in a therapeutic
benefit to the patient
. In the most basic sense, drug design involves design of small molecules that are complementary in shape
and charge
to the biomolecular target to which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling
techniques. This type of modeling is often referred to as computer-aided drug design.
The phrase "drug design" is to some extent a misnomer
. What is really meant by drug design is ligand
design. Modeling techniques for prediction of binding affinity are reasonably successful. However there are many other properties such as bioavailability
, metabolic half-life
, lack of side effects
, etc. that first must be optimized before a ligand can become a safe and efficacious drug. These other characteristics are often difficult to optimize using rational drug design techniques.
involved in a particular metabolic
or signaling
pathway that is specific to a disease condition or pathology
, or to the infectivity
or survival of a microbial
pathogen
. Some approaches attempt to inhibit the functioning of the pathway in the diseased state by causing a key molecule to stop functioning. Drugs may be designed that bind to the active region and inhibit this key molecule. Another approach may be to enhance the normal pathway by promoting specific molecules in the normal pathways that may have been affected in the diseased state. In addition, these drugs should also be designed in such a way as not to affect any other important "off-target" molecules or antitarget
s that may be similar in appearance to the target molecule, since drug interactions with off-target molecules may lead to undesirable side effect
s. Sequence homology is often used to identify such risks.
Most commonly, drugs are organic
small molecule
s produced through chemical synthesis, but biopolymer-based drugs (also known as biologics
) produced through biological processes are becoming increasingly more common. In addition, mRNA-based gene silencing
technologies may have therapeutic applications.
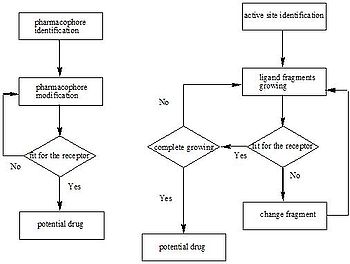 There are two major types of drug design. The first is referred to as ligand-based drug design and the second, structure-based drug design.
There are two major types of drug design. The first is referred to as ligand-based drug design and the second, structure-based drug design.
model that defines the minimum necessary structural characteristics a molecule must possess in order to bind to the target. In other words, a model of the biological target may be built based on the knowledge of what binds to it and this model in turn may be used to design new molecular entities that interact with the target. Alternatively, a quantitative structure-activity relationship
(QSAR) in which a correlation between calculated properties of molecules and their experimentally determined biological activity may be derived. These QSAR relationships in turn may be used to predict the activity of new analogs.
of the biological target obtained through methods such as x-ray crystallography or NMR spectroscopy
. If an experimental structure of a target is not available, it may be possible to create a homology model
of the target based on the experimental structure of a related protein. Using the structure of the biological target, candidate drugs that are predicted to bind with high affinity
and selectivity to the target may be designed using interactive graphics and the intuition of a medicinal chemist
. Alternatively various automated computational procedures may be used to suggest new drug candidates.
As experimental methods such as X-ray crystallography and NMR develop, the amount of information concerning 3D structures of biomolecular targets has increased dramatically. In parallel, information about the structural dynamics and electronic properties about ligands has also increased. This has encouraged the rapid development of the structure-based drug design. Current methods for structure-based drug design can be divided roughly into two categories. The first category is about “finding” ligands for a given receptor, which is usually referred as database searching. In this case, a large number of potential ligand molecules are screened to find those fitting the binding pocket of the receptor. This method is usually referred as ligand-based drug design. The key advantage of database searching is that it saves synthetic effort to obtain new lead compounds. Another category of structure-based drug design methods is about “building” ligands, which is usually referred as receptor-based drug design. In this case, ligand molecules are built up within the constraints of the binding pocket by assembling small pieces in a stepwise manner. These pieces can be either individual atoms or molecular fragments. The key advantage of such a method is that novel structures, not contained in any database, can be suggested. These techniques are raising much excitement to the drug design community.
The space inside the ligand binding region would be studied with virtual probe atoms of the four types above so the chemical environment of all spots in the ligand binding region can be known. Hence we are clear what kind of chemical fragments can be put into their corresponding spots in the ligand binding region of the receptor.
Before the first fragment, i.e. the seed, is put into the binding pocket, and other fragments can be added one by one, it is useful to identify potential problems. First, the possibility for the fragment combinations is huge. A small perturbation of the previous fragment conformation would cause great difference in the following construction process. At the same time, in order to find the lowest binding energy on the Potential energy surface
(PES) between planted fragments and receptor pocket, the scoring function calculation would be done for every step of conformation change of the fragments derived from every type of possible fragments combination. Since this requires a large amount of computation, one may think using other possible strategies to let the program works more efficiently. When a ligand is inserted into the pocket site of a receptor, conformation favor for these groups on the ligand that can bind tightly with receptor should be taken priority. Therefore it allows us to put several seeds at the same time into the regions that have significant interactions with the seeds and adjust their favorite conformation first, and then connect those seeds into a continuous ligand in a manner that make the rest part of the ligand having the lowest energy. The conformations of the pre-placed seeds ensuring the binding affinity decide the manner that ligand would be grown. This strategy reduces calculation burden for the fragment construction efficiently. On the other hand, it reduces the possibility of the combination of fragments, which reduces the number of possible ligands that can be derived from the program. These two strategies above are well used in most structure-based drug design programs. They are described as “Grow” and “Link”. The two strategies are always combined in order to make the construction result more reliable.
A breakthrough work was done by Böhm to develop a general-purposed empirical function in order to describe the binding energy.

The concept of the “Master Equation” was raised. The basic idea is that the overall binding free energy can be decomposed into independent components that are known to be important for the binding process. Each component reflects a certain kind of free energy alteration during the binding process between a ligand and its target receptor. The Master Equation is the linear combination of these components. According to Gibbs free energy equation, the relation between dissociation equilibrium constant, Kd, and the components of free energy alternation was built.
The sub models of empirical functions differ due to the consideration of researchers. It has long been a scientific challenge to design the sub models. Depending on the modification of them, the empirical scoring function is improved and continuously consummated.
, which rely on trial-and-error testing of chemical substances on cultured cell
s or animal
s, and matching the apparent effects to treatments, rational drug design begins with a hypothesis that modulation of a specific biological target may have therapeutic value. In order for a biomolecule to be selected as a drug target, two essential pieces of information are required. The first is evidence that modulation of the target will have therapeutic value. This knowledge may come from, for example, disease linkage studies that show an association between mutations in the biological target and certain disease states. The second is that the target is "drugable". This means that it is capable of binding to a small molecule and that its activity can be modulated by the small molecule.
Once a suitable target has been identified, the target is normally cloned
and expressed
. The expressed target is then used to establish a screening assay. In addition, the three-dimensional structure of the target may be determined.
The search for small molecules that bind to the target is begun by screening libraries of potential drug compounds. This may be done by using the screening assay (a "wet screen"). In addition, if the structure of the target is available, a virtual screen
may be performed of candidate drugs. Ideally the candidate drug compounds should be "drug-like
", that is they should possess properties that are predicted to lead to oral bioavailability, adequate chemical and metabolic stability, and minimal toxic effects. Several methods are available to estimate druglikeness such Lipinski's Rule of Five
and a range of scoring methods such as Lipophilic efficiency
. Several methods for predicting drug metabolism have been proposed in the scientific literature, and a recent example is SPORCalc. Due to the complexity of the drug design process, two terms of interest are still serendipity and bounded rationality
. Those challenges are caused by the large chemical space
describing potential new drugs without side-effect
s.
to discover, enhance, or study drugs
and related biologically active molecule
s. The most fundamental goal is to predict whether a given molecule will bind to a target and if so how strongly. Molecular mechanics
or molecular dynamics
are most often used to predict the conformation of the small molecule
and to model conformational changes in the biological target that may occur when the small molecule binds to it. Semi-empirical, ab initio quantum chemistry methods
, or density functional theory
are often used to provide optimized parameters for the molecular mechanics calculations and also provide an estimate of the electronic properties (electrostatic potential, polarizability
, etc.) of the drug candidate that will influence binding affinity.
Molecular mechanics methods may also be used to provide semi-quantitative prediction of the binding affinity. Also, knowledge-based scoring function
may be used to provide binding affinity estimates. These methods use linear regression
, machine learning
, neural nets or other statistical techniques to derive predictive binding affinity equations by fitting experimental affinities to computationally derived interaction energies between the small molecule and the target.
Ideally the computational method should be able to predict affinity before a compound is synthesized and hence in theory only one compound needs to be synthesized. The reality however is that present computational methods provide at best only qualitative accurate estimates of affinity. Therefore in practice it still takes several iterations of design, synthesis, and testing before an optimal molecule is discovered. On the other hand, computational methods have accelerated discovery by reducing the number of iterations required and in addition have often provided more novel small molecule structures.
Drug design with the help of computers may be used at any of the following stages of drug discovery:
In order to overcome the insufficient prediction of binding affinity calculated by recent scoring functions, the protein-ligand interaction and compound 3D structure information are used to analysis. For structure-based drug design, several post-screening analysis focusing on protein-ligand interaction has been developed for improving enrichment and effectively mining potential candidates:
, which was approved in 1995.
Another important case study in rational drug design is imatinib
, a tyrosine kinase
inhibitor designed specifically for the bcr-abl fusion protein that is characteristic for Philadelphia chromosome
-positive leukemia
s (chronic myelogenous leukemia
and occasionally acute lymphocytic leukemia). Imatinib is substantially different from previous drugs for cancer
, as most agents of chemotherapy
simply target rapidly dividing cells, not differentiating between cancer cells and other tissues.
Additional examples include:
Case studies
Rational design
In chemical biology and biomolecular engineering, rational design is the strategy of creating new molecules with a certain functionality, based upon the ability to predict how the molecule's structure will affect its behavior through physical models...
drug design or structure-based drug design, is the inventive
Invention
An invention is a novel composition, device, or process. An invention may be derived from a pre-existing model or idea, or it could be independently conceived, in which case it may be a radical breakthrough. In addition, there is cultural invention, which is an innovative set of useful social...
process of finding new medications based on the knowledge of the biological target
Biological target
A biological target is a biopolymer such as a protein or nucleic acid whose activity can be modified by an external stimulus. The definition is context-dependent and can refer to the biological target of a pharmacologically active drug compound, or the receptor target of a hormone . The...
. The drug is most commonly an organic
Organic compound
An organic compound is any member of a large class of gaseous, liquid, or solid chemical compounds whose molecules contain carbon. For historical reasons discussed below, a few types of carbon-containing compounds such as carbides, carbonates, simple oxides of carbon, and cyanides, as well as the...
small molecule
Small molecule
In the fields of pharmacology and biochemistry, a small molecule is a low molecular weight organic compound which is by definition not a polymer...
that activates or inhibits the function of a biomolecule
Biomolecule
A biomolecule is any molecule that is produced by a living organism, including large polymeric molecules such as proteins, polysaccharides, lipids, and nucleic acids as well as small molecules such as primary metabolites, secondary metabolites, and natural products...
such as a protein
Protein
Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form, facilitating a biological function. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of...
, which in turn results in a therapeutic
Therapeutic effect
A therapeutic effect is a consequence of a medical treatment of any kind, the results of which are judged to be desirable and beneficial. This is true whether the result was expected, unexpected, or even an unintended consequence of the treatment...
benefit to the patient
Patient
A patient is any recipient of healthcare services. The patient is most often ill or injured and in need of treatment by a physician, advanced practice registered nurse, veterinarian, or other health care provider....
. In the most basic sense, drug design involves design of small molecules that are complementary in shape
Shape
The shape of an object located in some space is a geometrical description of the part of that space occupied by the object, as determined by its external boundary – abstracting from location and orientation in space, size, and other properties such as colour, content, and material...
and charge
Electric charge
Electric charge is a physical property of matter that causes it to experience a force when near other electrically charged matter. Electric charge comes in two types, called positive and negative. Two positively charged substances, or objects, experience a mutual repulsive force, as do two...
to the biomolecular target to which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling
Molecular modelling
Molecular modelling encompasses all theoretical methods and computational techniques used to model or mimic the behaviour of molecules. The techniques are used in the fields of computational chemistry, computational biology and materials science for studying molecular systems ranging from small...
techniques. This type of modeling is often referred to as computer-aided drug design.
The phrase "drug design" is to some extent a misnomer
Misnomer
A misnomer is a term which suggests an interpretation that is known to be untrue. Such incorrect terms sometimes derive their names because of the form, action, or origin of the subject becoming named popularly or widely referenced—long before their true natures were known.- Sources of misnomers...
. What is really meant by drug design is ligand
Ligand (biochemistry)
In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. In a narrower sense, it is a signal triggering molecule, binding to a site on a target protein.The binding occurs by intermolecular forces, such as ionic bonds, hydrogen...
design. Modeling techniques for prediction of binding affinity are reasonably successful. However there are many other properties such as bioavailability
Bioavailability
In pharmacology, bioavailability is a subcategory of absorption and is used to describe the fraction of an administered dose of unchanged drug that reaches the systemic circulation, one of the principal pharmacokinetic properties of drugs. By definition, when a medication is administered...
, metabolic half-life
Biological half-life
The biological half-life or elimination half-life of a substance is the time it takes for a substance to lose half of its pharmacologic, physiologic, or radiologic activity, as per the MeSH definition...
, lack of side effects
Adverse drug reaction
An adverse drug reaction is an expression that describes harm associated with the use of given medications at a normal dosage. ADRs may occur following a single dose or prolonged administration of a drug or result from the combination of two or more drugs...
, etc. that first must be optimized before a ligand can become a safe and efficacious drug. These other characteristics are often difficult to optimize using rational drug design techniques.
Background
Typically a drug target is a key moleculeMolecule
A molecule is an electrically neutral group of at least two atoms held together by covalent chemical bonds. Molecules are distinguished from ions by their electrical charge...
involved in a particular metabolic
Metabolic pathway
In biochemistry, metabolic pathways are series of chemical reactions occurring within a cell. In each pathway, a principal chemical is modified by a series of chemical reactions. Enzymes catalyze these reactions, and often require dietary minerals, vitamins, and other cofactors in order to function...
or signaling
Signal transduction
Signal transduction occurs when an extracellular signaling molecule activates a cell surface receptor. In turn, this receptor alters intracellular molecules creating a response...
pathway that is specific to a disease condition or pathology
Pathology
Pathology is the precise study and diagnosis of disease. The word pathology is from Ancient Greek , pathos, "feeling, suffering"; and , -logia, "the study of". Pathologization, to pathologize, refers to the process of defining a condition or behavior as pathological, e.g. pathological gambling....
, or to the infectivity
Infectivity
In epidemiology, infectivity refers to the ability of a pathogen to establish an infection. More specifically, infectivity is a pathogen's capacity for horizontal transmission that is, how frequently it spreads among hosts that are not in a parent-child relationship...
or survival of a microbial
Microorganism
A microorganism or microbe is a microscopic organism that comprises either a single cell , cell clusters, or no cell at all...
pathogen
Pathogen
A pathogen gignomai "I give birth to") or infectious agent — colloquially, a germ — is a microbe or microorganism such as a virus, bacterium, prion, or fungus that causes disease in its animal or plant host...
. Some approaches attempt to inhibit the functioning of the pathway in the diseased state by causing a key molecule to stop functioning. Drugs may be designed that bind to the active region and inhibit this key molecule. Another approach may be to enhance the normal pathway by promoting specific molecules in the normal pathways that may have been affected in the diseased state. In addition, these drugs should also be designed in such a way as not to affect any other important "off-target" molecules or antitarget
Antitarget
In pharmacology, an antitarget is a receptor, enzyme, or other biological target that, when affected by a drug, causes undesirable side-effects. During drug design and development, it is important for pharmaceutical companies to ensure that new drugs do not show significant activity at any of a...
s that may be similar in appearance to the target molecule, since drug interactions with off-target molecules may lead to undesirable side effect
Adverse effect
In medicine, an adverse effect is a harmful and undesired effect resulting from a medication or other intervention such as surgery.An adverse effect may be termed a "side effect", when judged to be secondary to a main or therapeutic effect. If it results from an unsuitable or incorrect dosage or...
s. Sequence homology is often used to identify such risks.
Most commonly, drugs are organic
Organic compound
An organic compound is any member of a large class of gaseous, liquid, or solid chemical compounds whose molecules contain carbon. For historical reasons discussed below, a few types of carbon-containing compounds such as carbides, carbonates, simple oxides of carbon, and cyanides, as well as the...
small molecule
Small molecule
In the fields of pharmacology and biochemistry, a small molecule is a low molecular weight organic compound which is by definition not a polymer...
s produced through chemical synthesis, but biopolymer-based drugs (also known as biologics
Biologics
A biologic is a medicinal product such as a vaccine, blood or blood component, allergenic, somatic cell, gene therapy, tissue, recombinant therapeutic protein, or living cells that are used as therapeutics to treat diseases...
) produced through biological processes are becoming increasingly more common. In addition, mRNA-based gene silencing
Gene silencing
Gene silencing is a general term describing epigenetic processes of gene regulation. The term gene silencing is generally used to describe the "switching off" of a gene by a mechanism other than genetic modification...
technologies may have therapeutic applications.
Types
Ligand-based
Ligand-based drug design (or indirect drug design) relies on knowledge of other molecules that bind to the biological target of interest. These other molecules may be used to derive a pharmacophorePharmacophore
thumb|right|300px|An example of a pharmacophore model.A pharmacophore is an abstract description of molecular features which are necessary for molecular recognition of a ligand by a biological macromolecule....
model that defines the minimum necessary structural characteristics a molecule must possess in order to bind to the target. In other words, a model of the biological target may be built based on the knowledge of what binds to it and this model in turn may be used to design new molecular entities that interact with the target. Alternatively, a quantitative structure-activity relationship
Quantitative structure-activity relationship
Quantitative structure–activity relationship or QSPR is the process by which chemical structure is quantitatively correlated with a well defined process, such as biological activity or chemical reactivity.For example, biological activity can be expressed quantitatively as the concentration of a...
(QSAR) in which a correlation between calculated properties of molecules and their experimentally determined biological activity may be derived. These QSAR relationships in turn may be used to predict the activity of new analogs.
Structure-based
Structure-based drug design (or direct drug design) relies on knowledge of the three dimensional structureTertiary structure
In biochemistry and molecular biology, the tertiary structure of a protein or any other macromolecule is its three-dimensional structure, as defined by the atomic coordinates.-Relationship to primary structure:...
of the biological target obtained through methods such as x-ray crystallography or NMR spectroscopy
Protein nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance spectroscopy of proteins is a field of structural biology in which NMR spectroscopy is used to obtain information about the structure and dynamics of proteins. The field was pioneered by Richard R. Ernst and Kurt Wüthrich, among others...
. If an experimental structure of a target is not available, it may be possible to create a homology model
Homology modeling
Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein...
of the target based on the experimental structure of a related protein. Using the structure of the biological target, candidate drugs that are predicted to bind with high affinity
Dissociation constant
In chemistry, biochemistry, and pharmacology, a dissociation constant is a specific type of equilibrium constant that measures the propensity of a larger object to separate reversibly into smaller components, as when a complex falls apart into its component molecules, or when a salt splits up into...
and selectivity to the target may be designed using interactive graphics and the intuition of a medicinal chemist
Medicinal chemistry
Medicinal chemistry and pharmaceutical chemistry are disciplines at the intersection of chemistry, especially synthetic organic chemistry, and pharmacology and various other biological specialties, where it is involved with design, chemical synthesis and development for market of pharmaceutical...
. Alternatively various automated computational procedures may be used to suggest new drug candidates.
As experimental methods such as X-ray crystallography and NMR develop, the amount of information concerning 3D structures of biomolecular targets has increased dramatically. In parallel, information about the structural dynamics and electronic properties about ligands has also increased. This has encouraged the rapid development of the structure-based drug design. Current methods for structure-based drug design can be divided roughly into two categories. The first category is about “finding” ligands for a given receptor, which is usually referred as database searching. In this case, a large number of potential ligand molecules are screened to find those fitting the binding pocket of the receptor. This method is usually referred as ligand-based drug design. The key advantage of database searching is that it saves synthetic effort to obtain new lead compounds. Another category of structure-based drug design methods is about “building” ligands, which is usually referred as receptor-based drug design. In this case, ligand molecules are built up within the constraints of the binding pocket by assembling small pieces in a stepwise manner. These pieces can be either individual atoms or molecular fragments. The key advantage of such a method is that novel structures, not contained in any database, can be suggested. These techniques are raising much excitement to the drug design community.
Active site identification
Active site identification is the first step in this program. It analyzes the protein to find the binding pocket, derives key interaction sites within the binding pocket, and then prepares the necessary data for Ligand fragment link. The basic inputs for this step are the 3D structure of the protein and a pre-docked ligand in PDB format, as well as their atomic properties. Both ligand and protein atoms need to be classified and their atomic properties should be defined, basically, into four atomic types:- hydrophobic atom: All carbons in hydrocarbon chains or in aromatic groups.
- H-bond donor: Oxygen and nitrogen atoms bonded to hydrogen atom(s).
- H-bond acceptor: Oxygen and sp2 or sp hybridized nitrogen atoms with lone electron pair(s).
- Polar atom: Oxygen and nitrogen atoms that are neither H-bond donor nor H-bond acceptor, sulfur, phosphorus, halogen, metal, and carbon atoms bonded to hetero-atom(s).
The space inside the ligand binding region would be studied with virtual probe atoms of the four types above so the chemical environment of all spots in the ligand binding region can be known. Hence we are clear what kind of chemical fragments can be put into their corresponding spots in the ligand binding region of the receptor.
Ligand fragment link
When we want to plant “seeds” into different regions defined by the previous section, we need a fragments database to choose fragments from. The term “fragment” is used here to describe the building blocks used in the construction process. The rationale of this algorithm lies in the fact that organic structures can be decomposed into basic chemical fragments. Although the diversity of organic structures is infinite, the number of basic fragments is rather limited.Before the first fragment, i.e. the seed, is put into the binding pocket, and other fragments can be added one by one, it is useful to identify potential problems. First, the possibility for the fragment combinations is huge. A small perturbation of the previous fragment conformation would cause great difference in the following construction process. At the same time, in order to find the lowest binding energy on the Potential energy surface
Potential energy surface
A potential energy surface is generally used within the adiabatic or Born–Oppenheimer approximation in quantum mechanics and statistical mechanics to model chemical reactions and interactions in simple chemical and physical systems...
(PES) between planted fragments and receptor pocket, the scoring function calculation would be done for every step of conformation change of the fragments derived from every type of possible fragments combination. Since this requires a large amount of computation, one may think using other possible strategies to let the program works more efficiently. When a ligand is inserted into the pocket site of a receptor, conformation favor for these groups on the ligand that can bind tightly with receptor should be taken priority. Therefore it allows us to put several seeds at the same time into the regions that have significant interactions with the seeds and adjust their favorite conformation first, and then connect those seeds into a continuous ligand in a manner that make the rest part of the ligand having the lowest energy. The conformations of the pre-placed seeds ensuring the binding affinity decide the manner that ligand would be grown. This strategy reduces calculation burden for the fragment construction efficiently. On the other hand, it reduces the possibility of the combination of fragments, which reduces the number of possible ligands that can be derived from the program. These two strategies above are well used in most structure-based drug design programs. They are described as “Grow” and “Link”. The two strategies are always combined in order to make the construction result more reliable.
Scoring method
Structure-based drug design attempts to use the structure of proteins as a basis for designing new ligands by applying accepted principles of molecular recognition. The basic assumption underlying structure-based drug design is that a good ligand molecule should bind tightly to its target. Thus, one of the most important principles for designing or obtaining potential new ligands is to predict the binding affinity of a certain ligand to its target and use it as a criterion for selection.A breakthrough work was done by Böhm to develop a general-purposed empirical function in order to describe the binding energy.
The concept of the “Master Equation” was raised. The basic idea is that the overall binding free energy can be decomposed into independent components that are known to be important for the binding process. Each component reflects a certain kind of free energy alteration during the binding process between a ligand and its target receptor. The Master Equation is the linear combination of these components. According to Gibbs free energy equation, the relation between dissociation equilibrium constant, Kd, and the components of free energy alternation was built.
The sub models of empirical functions differ due to the consideration of researchers. It has long been a scientific challenge to design the sub models. Depending on the modification of them, the empirical scoring function is improved and continuously consummated.
Rational drug discovery
In contrast to traditional methods of drug discoveryDrug discovery
In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which drugs are discovered or designed.In the past most drugs have been discovered either by identifying the active ingredient from traditional remedies or by serendipitous discovery...
, which rely on trial-and-error testing of chemical substances on cultured cell
Cell culture
Cell culture is the complex process by which cells are grown under controlled conditions. In practice, the term "cell culture" has come to refer to the culturing of cells derived from singlecellular eukaryotes, especially animal cells. However, there are also cultures of plants, fungi and microbes,...
s or animal
Animal
Animals are a major group of multicellular, eukaryotic organisms of the kingdom Animalia or Metazoa. Their body plan eventually becomes fixed as they develop, although some undergo a process of metamorphosis later on in their life. Most animals are motile, meaning they can move spontaneously and...
s, and matching the apparent effects to treatments, rational drug design begins with a hypothesis that modulation of a specific biological target may have therapeutic value. In order for a biomolecule to be selected as a drug target, two essential pieces of information are required. The first is evidence that modulation of the target will have therapeutic value. This knowledge may come from, for example, disease linkage studies that show an association between mutations in the biological target and certain disease states. The second is that the target is "drugable". This means that it is capable of binding to a small molecule and that its activity can be modulated by the small molecule.
Once a suitable target has been identified, the target is normally cloned
Molecular cloning
Molecular cloning refers to a set of experimental methods in molecular biology that are used to assemble recombinant DNA molecules and to direct their replication within host organisms...
and expressed
Protein expression
Protein expression is a subcomponent of gene expression. It consists of the stages after DNA has been translated into polypeptide chains, which are ultimately folded into proteins...
. The expressed target is then used to establish a screening assay. In addition, the three-dimensional structure of the target may be determined.
The search for small molecules that bind to the target is begun by screening libraries of potential drug compounds. This may be done by using the screening assay (a "wet screen"). In addition, if the structure of the target is available, a virtual screen
Virtual screening
Virtual screening is a computational technique used in drug discovery research. By using computers, it deals with the quick search of large libraries of chemical structures in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or...
may be performed of candidate drugs. Ideally the candidate drug compounds should be "drug-like
Druglikeness
Druglikeness is a qualitative concept used in drug design for how "druglike" a substance is with respect to factors like bioavailability. It is estimated from the molecular structure before the substance is even synthesized and tested...
", that is they should possess properties that are predicted to lead to oral bioavailability, adequate chemical and metabolic stability, and minimal toxic effects. Several methods are available to estimate druglikeness such Lipinski's Rule of Five
Lipinski's Rule of Five
Lipinski's Rule of Five is a rule of thumb to evaluate druglikeness or determine if a chemical compound with a certain pharmacological or biological activity has properties that would make it a likely orally active drug in humans. The rule was formulated by Christopher A...
and a range of scoring methods such as Lipophilic efficiency
Lipophilic efficiency
Lipophilic efficiency , sometimes referred toas ligand-lipophilicity efficiency is a parameter used in drug design and drug discovery to evaluate the quality of research compounds, linking potency and lipophilicity in an attempt to estimate druglikeness...
. Several methods for predicting drug metabolism have been proposed in the scientific literature, and a recent example is SPORCalc. Due to the complexity of the drug design process, two terms of interest are still serendipity and bounded rationality
Bounded rationality
Bounded rationality is the idea that in decision making, rationality of individuals is limited by the information they have, the cognitive limitations of their minds, and the finite amount of time they have to make a decision...
. Those challenges are caused by the large chemical space
Chemical space
Chemical space is the space spanned by all possible molecules and chemical compounds – that is, all stoichiometric combinations of electrons and atomic nuclei, in all possible topology isomers. Chemical reactions allow us to move in chemical space...
describing potential new drugs without side-effect
Adverse effect
In medicine, an adverse effect is a harmful and undesired effect resulting from a medication or other intervention such as surgery.An adverse effect may be termed a "side effect", when judged to be secondary to a main or therapeutic effect. If it results from an unsuitable or incorrect dosage or...
s.
Computer-assisted drug design
Computer-assisted drug design uses computational chemistryComputational chemistry
Computational chemistry is a branch of chemistry that uses principles of computer science to assist in solving chemical problems. It uses the results of theoretical chemistry, incorporated into efficient computer programs, to calculate the structures and properties of molecules and solids...
to discover, enhance, or study drugs
DRUGS
Destroy Rebuild Until God Shows are an American post-hardcore band formed in 2010. They released their debut self-titled album on February 22, 2011.- Formation :...
and related biologically active molecule
Molecule
A molecule is an electrically neutral group of at least two atoms held together by covalent chemical bonds. Molecules are distinguished from ions by their electrical charge...
s. The most fundamental goal is to predict whether a given molecule will bind to a target and if so how strongly. Molecular mechanics
Molecular mechanics
Molecular mechanics uses Newtonian mechanics to model molecular systems. The potential energy of all systems in molecular mechanics is calculated using force fields...
or molecular dynamics
Molecular dynamics
Molecular dynamics is a computer simulation of physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a period of time, giving a view of the motion of the atoms...
are most often used to predict the conformation of the small molecule
Small molecule
In the fields of pharmacology and biochemistry, a small molecule is a low molecular weight organic compound which is by definition not a polymer...
and to model conformational changes in the biological target that may occur when the small molecule binds to it. Semi-empirical, ab initio quantum chemistry methods
Ab initio quantum chemistry methods
Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initiowas first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited states of benzene.The background is described by Parr...
, or density functional theory
Density functional theory
Density functional theory is a quantum mechanical modelling method used in physics and chemistry to investigate the electronic structure of many-body systems, in particular atoms, molecules, and the condensed phases. With this theory, the properties of a many-electron system can be determined by...
are often used to provide optimized parameters for the molecular mechanics calculations and also provide an estimate of the electronic properties (electrostatic potential, polarizability
Polarizability
Polarizability is the measure of the change in a molecule's electron distribution in response to an applied electric field, which can also be induced by electric interactions with solvents or ionic reagents. It is a property of matter...
, etc.) of the drug candidate that will influence binding affinity.
Molecular mechanics methods may also be used to provide semi-quantitative prediction of the binding affinity. Also, knowledge-based scoring function
Scoring functions for docking
In the fields of computational chemistry and molecular modelling, scoring functions are fast approximate mathematical methods used to predict the strength of the non-covalent interaction between two molecules after they have been docked...
may be used to provide binding affinity estimates. These methods use linear regression
Linear regression
In statistics, linear regression is an approach to modeling the relationship between a scalar variable y and one or more explanatory variables denoted X. The case of one explanatory variable is called simple regression...
, machine learning
Machine learning
Machine learning, a branch of artificial intelligence, is a scientific discipline concerned with the design and development of algorithms that allow computers to evolve behaviors based on empirical data, such as from sensor data or databases...
, neural nets or other statistical techniques to derive predictive binding affinity equations by fitting experimental affinities to computationally derived interaction energies between the small molecule and the target.
Ideally the computational method should be able to predict affinity before a compound is synthesized and hence in theory only one compound needs to be synthesized. The reality however is that present computational methods provide at best only qualitative accurate estimates of affinity. Therefore in practice it still takes several iterations of design, synthesis, and testing before an optimal molecule is discovered. On the other hand, computational methods have accelerated discovery by reducing the number of iterations required and in addition have often provided more novel small molecule structures.
Drug design with the help of computers may be used at any of the following stages of drug discovery:
- hit identification using virtual screeningVirtual screeningVirtual screening is a computational technique used in drug discovery research. By using computers, it deals with the quick search of large libraries of chemical structures in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or...
(structure- or ligand-based design) - hit-to-leadDrug discovery hit to leadEarly drug discovery involves several phases from target identification to preclinical development. The identification of small molecule modulators of protein function and the process of transforming these into high-content lead series are key activities in modern drug discovery. The Hit-to-Lead...
optimization of affinity and selectivity (structure-based design, QSARQuantitative structure-activity relationshipQuantitative structure–activity relationship or QSPR is the process by which chemical structure is quantitatively correlated with a well defined process, such as biological activity or chemical reactivity.For example, biological activity can be expressed quantitatively as the concentration of a...
, etc.) - lead optimizationDrug developmentDrug development is a blanket term used to define the process of bringing a new drug to the market once a lead compound has been identified through the process of drug discovery...
optimization of other pharmaceutical properties while maintaining affinity
In order to overcome the insufficient prediction of binding affinity calculated by recent scoring functions, the protein-ligand interaction and compound 3D structure information are used to analysis. For structure-based drug design, several post-screening analysis focusing on protein-ligand interaction has been developed for improving enrichment and effectively mining potential candidates:
- Consensus scoring
- Selecting candidates by voting of multiple scoring functions
- May lose the relationship between protein-ligand structural information and scoring criterion
- Geometric analysis
- Comparing protein-ligand interactions by visually inspecting individual structures
- Becoming intractable when the number of complexes to be analyzed increasing
- Cluster analysis
- Represent and cluster candidates according to protein-ligand 3D information
- Needs meaningful representation of protein-ligand interactions.
Examples
A particular example of rational drug design involves the use of three-dimensional information about biomolecules obtained from such techniques as X-ray crystallography and NMR spectroscopy. This approach to drug discovery is sometimes referred to as structure-based drug design. The first unequivocal example of the application of structure-based drug design leading to an approved drug is the carbonic anhydrase inhibitor dorzolamideDorzolamide
Dorzolamide is a carbonic anhydrase inhibitor. It is an anti-glaucoma agent and topically applied in the form of eye drops. This drug, developed by Merck, was the first drug in human therapy which resulted from structure-based drug design...
, which was approved in 1995.
Another important case study in rational drug design is imatinib
Imatinib
Imatinib is a drug used to treat certain types of cancer. It is currently marketed by Novartis as Gleevec or Glivec as its mesylate salt, imatinib mesilate . It is used in treating chronic myelogenous leukemia , gastrointestinal stromal tumors and some other diseases...
, a tyrosine kinase
Tyrosine kinase
A tyrosine kinase is an enzyme that can transfer a phosphate group from ATP to a protein in a cell. It functions as an "on" or "off" switch in many cellular functions....
inhibitor designed specifically for the bcr-abl fusion protein that is characteristic for Philadelphia chromosome
Philadelphia chromosome
Philadelphia chromosome or Philadelphia translocation is a specific chromosomal abnormality that is associated with chronic myelogenous leukemia . It is the result of a reciprocal translocation between chromosome 9 and 22, and is specifically designated t...
-positive leukemia
Leukemia
Leukemia or leukaemia is a type of cancer of the blood or bone marrow characterized by an abnormal increase of immature white blood cells called "blasts". Leukemia is a broad term covering a spectrum of diseases...
s (chronic myelogenous leukemia
Chronic myelogenous leukemia
Chronic myelogenous leukemia , also known as chronic granulocytic leukemia , is a cancer of the white blood cells. It is a form of leukemia characterized by the increased and unregulated growth of predominantly myeloid cells in the bone marrow and the accumulation of these cells in the blood...
and occasionally acute lymphocytic leukemia). Imatinib is substantially different from previous drugs for cancer
Cancer
Cancer , known medically as a malignant neoplasm, is a large group of different diseases, all involving unregulated cell growth. In cancer, cells divide and grow uncontrollably, forming malignant tumors, and invade nearby parts of the body. The cancer may also spread to more distant parts of the...
, as most agents of chemotherapy
Chemotherapy
Chemotherapy is the treatment of cancer with an antineoplastic drug or with a combination of such drugs into a standardized treatment regimen....
simply target rapidly dividing cells, not differentiating between cancer cells and other tissues.
Additional examples include:
Case studies
- 5-HT3 antagonists
- Acetylcholine receptor agonists
- Angiotensin receptor blockersDiscovery and development of angiotensin receptor blockersThe angiotensin receptor blockers , also called angiotensin receptor antagonists or sartans, are a group of anti-hypertensive drugs that act by blocking the effects of the hormone angiotensin II in the body, thereby lowering blood pressure...
- Bcr-Abl tyrosine kinase inhibitors
- Cannabinoid receptor antagonists
- CCR5 receptor antagonistsDiscovery and development of CCR5 receptor antagonistsCCR5 receptor antagonists are a class of small molecules that antagonize the CCR5 receptor. The C-C motif chemokine receptor CCR5 is involved in the HIV entry process...
- Cyclooxygenase 2 inhibitors
- Dipeptidyl peptidase-4 inhibitorsDevelopment of dipeptidyl peptidase-4 inhibitorsDipeptidyl peptidase-4 inhibitors are enzyme inhibitors that inhibit the enzyme dipeptidyl peptidase-4 and are a potent treatment for type 2 diabetes...
- HIV protease inhibitorsDiscovery and development of HIV protease inhibitorsMany major physiological processes depend on regulation of proteolytic enzyme activity and there can be dramatic consequences when equilibrium between an enzyme and its substrates is disturbed. In this prospective, the discovery of small-molecule ligands, like protease inhibitors, that can modulate...
- NK1 receptor antagonists
- Non-nucleoside reverse transcriptase inhibitorsDiscovery and development of non-nucleoside reverse transcriptase inhibitorsNon-nucleoside reverse-transcriptase inhibitors are antiretroviral drugs used in the treatment of human immunodeficiency virus . NNRTIs inhibit reverse transcriptase , an enzyme that controls the replication of the genetic material of HIV...
- Proton pump inibitorsDiscovery and development of proton pump inhibitorsProton pump inhibitors block the gastric Hydrogen potassium ATPase and inhibit gastric acid secretion. These drugs have emerged as the treatment of choice for acid-related diseases, including gastroesophageal reflux disease and peptic ulcer disease.-History:Evidence emerged by the end of the...
- Triptans
- TRPV1 antagonistsDiscovery and development of TRPV1 antagonistsChronic pain remains a recognized unmet medical need. Consequently, the search for new analgesic agents is being intensively studied by the pharmaceutical industry. The TRPV1 receptor is an ion channel that has been implicated in mediation of many types of pain and therefore studied most extensively...
- Renin inhibitorsDiscovery and development of renin inhibitorsRenin inhibitors are antihypertensive drugs that inhibit the first and rate-limiting step of the renin-angiotensin-aldosterone system . Since the 1970s scientists have been trying to develop potent inhibitors with acceptable oral bioavailability. The process was difficult and took about three decades...
- Discovery and development of small molecule c-Met inhibitorsDiscovery and development of small molecule c-Met inhibitorsThe c-Met tyrosine kinase stimulates cell scattering, invasion, protection from apoptosis and angiogenesis.c-Met is a receptor tyrosine kinase, which can cause a wide variety of different cancers, such as renal, gastric and small cell lung carcinomas, central nervous system tumours, as well as...
See also
External links
- Click2Drug.org - Directory of computational drug design tools.