

Human mitochondrial genetics
Encyclopedia
Human mitochondrial genetics is the study of the genetics
of the DNA
contained in human mitochondria. Mitochondria are small structures in cells that generate energy for the cell to use, and are hence referred to as the "powerhouses" of the cell.
Mitochondrial DNA
(mtDNA) is not transmitted through nuclear DNA
(nDNA). In humans, as in most multicellular organisms, mitochondrial DNA is inherited only from the mother's ovum
.
Mitochondrial inheritance is therefore non-Mendelian
, as Mendelian inheritance
presumes that half the genetic material of a fertilized egg (zygote
) derives from each parent.
Eighty percent of mitochondrial DNA codes
for functional mitochondrial proteins, and therefore most mitochondrial DNA mutations lead to functional problems, which may be manifested as muscle disorders (myopathies
).
Understanding the genetic mutations that affect mitochondria can help us to understand the inner workings of cells and organisms, as well as helping to suggest methods for successful therapeutic tissue and organ cloning, and to treatments or possibly cures for many devastating muscular disorders.
, mitochondria are essential to all higher organisms for sustaining life. The mitochondrial disease
s are genetic disorder
s carried
in mitochondrial DNA, or nuclear DNA coding for mitochondrial components. Slight problems with any one of the numerous enzymes used by the mitochondria can be devastating to the cell, and in turn, to the organism.
are mediated by protein complexes (named Complexes I-V, DHO-QO, ETF-QO, and ANT). Complex I, or NADH : coenzyme Q oxidoreductase
, uses the energy in NADH to pump protons into the intermembrane space of the mitochondrion, pumping 2 protons per electron and passing 2 electrons via coenzyme Q
to complex III or coenzyme Q : cytochrome c oxidoreductase
. Complex II or succinate : coenzyme Q oxidoreductase
accepts energy from succinate produced in the citric acid
cycle and passes it via coenzyme Q to complex III. Complex III pumps 1 protons per electron and passes 1 electron via cytochrome c
to complex IV or Cytochrome C : O2 Oxidoreductase
. Complex IV pumps 1 protons into the space between the mitochondrion’s two membranes before passing the electron to O2 which reacts to form water. Complex V (ATP synthase
) is a rotary complex which allows approximately (determining the actual number is very difficult) 10 protons to enter the mitochondrial matrix along their concentration gradients. It uses the energy from the gradient to form the bond between ADP and the phosphate group to create ATP. The electron transfer flavoprotein : coenzyme Q oxidoreductase is also an electron-transporting molecule and is involved in the breakdown of fatty acids and amino acids. ANT (adenine nucleotide translocator
) is also involved in oxidative phosphorylation
as an energy carrying molecule. Each of these eight complexes plays a vital role in the health of the cell and any slight mutation in any one of the proteins that make up these complexes can lead to cell death or stress, which can both in turn lead to a number of diseases.
(mtDNA) is present in mitochondria as a circular molecule and in most species codes for 13 or 14 proteins involved in the electron transfer chain, 2 rRNA subunits and 22 tRNA molecules (all necessary for protein synthesis). The number of proteins involved in the electron transfer chain is much larger than 13 or 14, but the others are coded by the nuclear DNA.
In total, the mitochondrion hosts about 3000 proteins, but only about 13 of them are coded on the mitochondrial DNA. Most of the 3000 proteins are involved in a variety of processes other than ATP production, such as porphyrin
synthesis. Only about 3% of them code for ATP production proteins. This means most of the genetic information coding for the protein makeup of mitochondria is in chromosomal DNA and is involved in processes other than ATP synthesis. This increases the chances that a mutation that will affect a mitochondrion will occur in chromosomal DNA, which is inherited in a Mendelian pattern. Another result is that a chromosomal mutation will affect a specific tissue due to its specific needs, whether those may be high energy requirements or a need for the catabolism or anabolism of a specific neurotransmitter or nucleic acid. Because several copies of the mitochondrial genome are carried by each mitochondrion (2-10 in humans), mitochondrial mutations can be inherited maternally by mtDNA mutations which are present in mitochondria inside the oocyte
before fertilization, or (as stated above) through mutations in the chromosomes.
In humans, the heavy strand of mtDNA carries 28 genes and the light strand of mtDNA carries only 9 genes. Eight of the 9 genes on the light strand code for mitochondrial tRNA molecules. Human mtDNA consists of 16,569 nucleotide pairs. The entire molecule is regulated by only one regulatory region which contains the origins of replication of both heavy and light strands. The entire human mitochondrial DNA molecule has been mapped. The rate of mutation
in mtDNA is calculated to be about ten times greater than that of nuclear DNA, possibly due to a paucity of DNA repair mechanisms. This high mutation rate leads to a high variation between mitochondria, not only among different species but even within the same species. It is calculated that if two humans are chosen randomly and their mtDNA is tested, they will have an average of between fifty and seventy different nucleotides. This may not seem like much, but when compared to the total number of nucleotides of a human mitochondrial DNA molecule (16,569), as much as 0.42% of the mtDNA varies between two people.
is, for the most part, universal, with few exceptions: mitochondrial genetics includes some of these. For most organisms the "stop codon
s" are “UAA”, “UAG”, and “UGA”. In vertebrate mitochondria “AGA” and “AGG” are also stop codons, but not “UGA”, which codes for tryptophan
instead. “AUA” codes for isoleucine
in most organisms but for methionine
in vertebrate mitochondrial mRNA.
There are many other variations among the codes used by other mitochondrial m/tRNA, which happened not to be harmful to their organisms, and which can be used as a tool (along with other mutations among the mtDNA/RNA of different species) to determine relative proximity of common ancestry of related species. (The more related two species are, the more mtDNA/RNA mutations will be the same in their mitochondrial genome).
Using these techniques, it is estimated that the first mitochondria arose around 1.5 billion years ago. A generally accepted hypothesis
is that mitochondria originated as an aerobic
prokaryote
in a symbiotic relationship
within an anaerobic
eukaryote
.
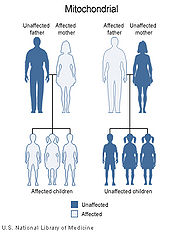 Because mitochondrial disease
Because mitochondrial disease
s (diseases due to malfunction of mitochondria) can be inherited both maternally and through chromosomal inheritance, the way in which they are passed on from generation to generation can vary greatly depending on the disease. Mitochondrial genetic mutations that occur in the nuclear DNA can occur in any of the chromosomes (depending on the species). Mutations inherited through the chromosomes can be autosomal dominant or recessive and can also be sex-linked dominant or recessive. Chromosomal inheritance follows normal Mendelian laws
, despite the fact that the phenotype of the disease may be masked.
Because of the complex ways in which mitochondrial and nuclear DNA "communicate" and interact, even seemingly simple inheritance is hard to diagnose. A mutation in chromosomal DNA may change a protein that regulates (increases or decreases) the production of another certain protein in the mitochondria or the cytoplasm; this may lead to slight, if any, noticeable symptoms. On the other hand, some devastating mtDNA mutations are easy to diagnose because of their widespread damage to muscular, neural, and/or hepatic tissues (among other high-energy and metabolism-dependent tissues) and because they are present in the mother and all the offspring.
Mitochondrial genome mutations are passed on 100% of the time from mother to all her offspring. The number of affected mtDNA molecules inherited by a specific offspring can vary greatly because
It is possible, even in twin births, for one baby to receive more than half mutant mtDNA molecules while the other twin may receive only a tiny fraction of mutant mtDNA molecules with respect to wildtype (depending on how the twins divide from each other and how many mutant mitochondria happen to be on each side of the division). In a few cases, some mitochondria or a mitochondrion from the sperm cell enters the oocyte but paternal mitochondria
are actively decomposed.
, or one long transcript. The production of primers occurs by processing of light strand transcripts with the Mitochondrial RNase MRP (Mitochondrial RNA Processing). The requirement of transcription to produce primers links the process of transcription to mtDNA replication. Full length transcripts are cut into functional tRNA, rRNA, and mRNA molecules. The process of transcription initiation in mitochondria involves three types of proteins: the mitochondrial RNA polymerase (POLRMT
), mitochondrial transcription factor A (TFAM), and mitochondrial transcription factors B1 and B2 (TFB1M, TFB2M). POLRMT
, TFAM, and TFB1M or TFB2M assemble at the mitochondrial promoters and begin transcription. The actual molecular events that are involved in initiation are unknown, but these factors make up the basal transcription machinery and have been shown to function in vitro. Mitochondrial translation is still not very well understood. In vitro
translations have still not been successful, probably due to the difficulty of isolating sufficient mt mRNA, functional mt rRNA, and possibly because of the complicated changes that the mRNA undergoes before it is translated.
gene) is used in the copying of mtDNA during replication. Because the two (heavy
and light) strands on the circular mtDNA molecule have different origins of replication
, it replicates in a D-loop mode
. One strand begins to replicate first, displacing the other strand. This continues until replication reaches the origin of replication on the other strand, at which point the other strand begins replicating in the opposite direction. This results in two new mtDNA molecules. Each mitochondrion has several copies of the mtDNA molecule and the number of mtDNA molecules is a limiting factor in mitochondrial fission
. After the mitochondrion has enough mtDNA, membrane area, and membrane proteins, it can undergo fission (very similar to that which bacteria use) to become two mitochondria. Evidence suggests that mitochondria can also undergo fusion and exchange (in a form of crossover
) genetic material among each other. Mitochondria sometimes form large matrices in which fusion, fission, and protein exchanges are constantly occurring. mtDNA shared among mitochondria (despite the fact that they can undergo fusion).
, Alzheimer’s
, and coronary artery disease
.
s range in severity from asymptomatic to fatal, and are most commonly due to inherited rather than acquired mutations of mitochondrial DNA. A given mitochondrial mutation can cause various diseases depending on the severity of the problem in the mitochondria and the tissue the affected mitochondria are in. Conversely, several different mutations may present themselves as the same disease. This almost patient-specific characterization of mitochondrial diseases (see Personalized medicine
) makes them very hard to accurately recognize, diagnose and trace. Some diseases are observable at or even before birth (many causing death) while others do not show themselves until late adulthood (late-onset disorders). This is because the number of mutant versus wildtype mitochondria varies between cells and tissues, and is continuously changing. Because cells have multiple mitochondria, different mitochondria in the same cell can have different variations of the mtDNA. This condition is referred to as heteroplasmy
. When a certain tissue reaches a certain ratio of mutant versus wildtype mitochondria, a disease will present itself. The ratio varies from person to person and tissue to tissue (depending on its specific energy, oxygen, and metabolism requirements, and the effects of the specific mutation). Mitochondrial diseases are very numerous and different. Apart from diseases caused by abnormalities in mitochondrial DNA, many diseases are suspected to be associated in part by mitochondrial dysfunctions, such as diabetes mellitus
, forms of cancer
and cardiovascular disease
, lactic acidosis
, specific forms of myopathy
, osteoporosis
, Alzheimer's disease
, Parkinsons's disease, stroke
, Male infertility
and which are also believed to play a role in the aging process.
Genetics
Genetics , a discipline of biology, is the science of genes, heredity, and variation in living organisms....
of the DNA
DNA
Deoxyribonucleic acid is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms . The DNA segments that carry this genetic information are called genes, but other DNA sequences have structural purposes, or are involved in...
contained in human mitochondria. Mitochondria are small structures in cells that generate energy for the cell to use, and are hence referred to as the "powerhouses" of the cell.
Mitochondrial DNA
Mitochondrial DNA
Mitochondrial DNA is the DNA located in organelles called mitochondria, structures within eukaryotic cells that convert the chemical energy from food into a form that cells can use, adenosine triphosphate...
(mtDNA) is not transmitted through nuclear DNA
Nuclear DNA
Nuclear DNA, nuclear deoxyribonucleic acid , is DNA contained within a nucleus of eukaryotic organisms. In mammals and vertebrates, nuclear DNA encodes more of the genome than the mitochondrial DNA and is composed of information inherited from two parents, one male, and one female, rather than...
(nDNA). In humans, as in most multicellular organisms, mitochondrial DNA is inherited only from the mother's ovum
Ovum
An ovum is a haploid female reproductive cell or gamete. Both animals and embryophytes have ova. The term ovule is used for the young ovum of an animal, as well as the plant structure that carries the female gametophyte and egg cell and develops into a seed after fertilization...
.
Mitochondrial inheritance is therefore non-Mendelian
Non-mendelian inheritance
Non-Mendelian inheritance is a general term that refers to any pattern of inheritance in which traits do not segregate in accordance with Mendel’s laws. These laws describe the inheritance of traits linked to single genes on chromosomes in the nucleus. In Mendelian inheritance, each parent...
, as Mendelian inheritance
Mendelian inheritance
Mendelian inheritance is a scientific description of how hereditary characteristics are passed from parent organisms to their offspring; it underlies much of genetics...
presumes that half the genetic material of a fertilized egg (zygote
Zygote
A zygote , or zygocyte, is the initial cell formed when two gamete cells are joined by means of sexual reproduction. In multicellular organisms, it is the earliest developmental stage of the embryo...
) derives from each parent.
Eighty percent of mitochondrial DNA codes
Genetic code
The genetic code is the set of rules by which information encoded in genetic material is translated into proteins by living cells....
for functional mitochondrial proteins, and therefore most mitochondrial DNA mutations lead to functional problems, which may be manifested as muscle disorders (myopathies
Myopathy
In medicine, a myopathy is a muscular disease in which the muscle fibers do not function for any one of many reasons, resulting in muscular weakness. "Myopathy" simply means muscle disease...
).
Understanding the genetic mutations that affect mitochondria can help us to understand the inner workings of cells and organisms, as well as helping to suggest methods for successful therapeutic tissue and organ cloning, and to treatments or possibly cures for many devastating muscular disorders.
Mitochondrial function and genome
Because they provide 36 molecules of ATP per glucose molecule in contrast to the 2 ATP molecules produced by glycolysisGlycolysis
Glycolysis is the metabolic pathway that converts glucose C6H12O6, into pyruvate, CH3COCOO− + H+...
, mitochondria are essential to all higher organisms for sustaining life. The mitochondrial disease
Mitochondrial disease
Mitochondrial diseases are a group of disorders caused by dysfunctional mitochondria, the organelles that are the "powerhouses" of the cell. Mitochondria are found in every cell of the human body except red blood cells...
s are genetic disorder
Genetic disorder
A genetic disorder is an illness caused by abnormalities in genes or chromosomes, especially a condition that is present from before birth. Most genetic disorders are quite rare and affect one person in every several thousands or millions....
s carried
in mitochondrial DNA, or nuclear DNA coding for mitochondrial components. Slight problems with any one of the numerous enzymes used by the mitochondria can be devastating to the cell, and in turn, to the organism.
Membrane complexes
The processes carried out by the electron transport chainElectron transport chain
An electron transport chain couples electron transfer between an electron donor and an electron acceptor with the transfer of H+ ions across a membrane. The resulting electrochemical proton gradient is used to generate chemical energy in the form of adenosine triphosphate...
are mediated by protein complexes (named Complexes I-V, DHO-QO, ETF-QO, and ANT). Complex I, or NADH : coenzyme Q oxidoreductase
NADH dehydrogenase
NADH dehydrogenase is an enzyme located in the inner mitochondrial membrane that catalyzes the transfer of electrons from NADH to coenzyme Q...
, uses the energy in NADH to pump protons into the intermembrane space of the mitochondrion, pumping 2 protons per electron and passing 2 electrons via coenzyme Q
Coenzyme Q
Coenzyme Q10, also known as ubiquinone, ubidecarenone, coenzyme Q, and abbreviated at times to CoQ10 , CoQ, Q10, or Q, is a 1,4-benzoquinone, where Q refers to the quinone chemical group, and 10 refers to the number of isoprenyl chemical subunits in its tail.This oil-soluble, vitamin-like substance...
to complex III or coenzyme Q : cytochrome c oxidoreductase
Coenzyme Q - cytochrome c reductase
In enzymology, a ubiquinol—cytochrome-c reductase is an enzyme that catalyzes the chemical reactionThus, the two substrates of this enzyme are dihydroquinone and ferri- cytochrome c, whereas its 3 products are quinone , ferro- cytochrome c, and H+.This enzyme belongs to the family of...
. Complex II or succinate : coenzyme Q oxidoreductase
Succinate - coenzyme Q reductase
Succinate dehydrogenase or succinate-coenzyme Q reductase or Complex II is an enzyme complex, bound to the inner mitochondrial membrane of mammalian mitochondria and many bacterial cells...
accepts energy from succinate produced in the citric acid
Citric acid
Citric acid is a weak organic acid. It is a natural preservative/conservative and is also used to add an acidic, or sour, taste to foods and soft drinks...
cycle and passes it via coenzyme Q to complex III. Complex III pumps 1 protons per electron and passes 1 electron via cytochrome c
Cytochrome c
The Cytochrome complex, or cyt c is a small heme protein found loosely associated with the inner membrane of the mitochondrion. It belongs to the cytochrome c family of proteins. Cytochrome c is a highly soluble protein, unlike other cytochromes, with a solubility of about 100 g/L and is an...
to complex IV or Cytochrome C : O2 Oxidoreductase
Cytochrome c oxidase
The enzyme cytochrome c oxidase or Complex IV is a large transmembrane protein complex found in bacteria and the mitochondrion.It is the last enzyme in the respiratory electron transport chain of mitochondria located in the mitochondrial membrane...
. Complex IV pumps 1 protons into the space between the mitochondrion’s two membranes before passing the electron to O2 which reacts to form water. Complex V (ATP synthase
ATP synthase
right|thumb|300px|Molecular model of ATP synthase by X-ray diffraction methodATP synthase is an important enzyme that provides energy for the cell to use through the synthesis of adenosine triphosphate . ATP is the most commonly used "energy currency" of cells from most organisms...
) is a rotary complex which allows approximately (determining the actual number is very difficult) 10 protons to enter the mitochondrial matrix along their concentration gradients. It uses the energy from the gradient to form the bond between ADP and the phosphate group to create ATP. The electron transfer flavoprotein : coenzyme Q oxidoreductase is also an electron-transporting molecule and is involved in the breakdown of fatty acids and amino acids. ANT (adenine nucleotide translocator
Adenine nucleotide translocator
Adenine nucleotide translocator also known as the ADP/ATP translocator is a mitochondrial protein.- Function :ANT has long been thought to function asymmetrically as a homodimer of subunits in the inner mitochondrial membrane. The dimer was thought to be a gated pore through which ADP and ATP were...
) is also involved in oxidative phosphorylation
Oxidative phosphorylation
Oxidative phosphorylation is a metabolic pathway that uses energy released by the oxidation of nutrients to produce adenosine triphosphate . Although the many forms of life on earth use a range of different nutrients, almost all aerobic organisms carry out oxidative phosphorylation to produce ATP,...
as an energy carrying molecule. Each of these eight complexes plays a vital role in the health of the cell and any slight mutation in any one of the proteins that make up these complexes can lead to cell death or stress, which can both in turn lead to a number of diseases.
Genome
Mitochondrial DNAMitochondrial DNA
Mitochondrial DNA is the DNA located in organelles called mitochondria, structures within eukaryotic cells that convert the chemical energy from food into a form that cells can use, adenosine triphosphate...
(mtDNA) is present in mitochondria as a circular molecule and in most species codes for 13 or 14 proteins involved in the electron transfer chain, 2 rRNA subunits and 22 tRNA molecules (all necessary for protein synthesis). The number of proteins involved in the electron transfer chain is much larger than 13 or 14, but the others are coded by the nuclear DNA.
In total, the mitochondrion hosts about 3000 proteins, but only about 13 of them are coded on the mitochondrial DNA. Most of the 3000 proteins are involved in a variety of processes other than ATP production, such as porphyrin
Porphyrin
Porphyrins are a group of organic compounds, many naturally occurring. One of the best-known porphyrins is heme, the pigment in red blood cells; heme is a cofactor of the protein hemoglobin. Porphyrins are heterocyclic macrocycles composed of four modified pyrrole subunits interconnected at...
synthesis. Only about 3% of them code for ATP production proteins. This means most of the genetic information coding for the protein makeup of mitochondria is in chromosomal DNA and is involved in processes other than ATP synthesis. This increases the chances that a mutation that will affect a mitochondrion will occur in chromosomal DNA, which is inherited in a Mendelian pattern. Another result is that a chromosomal mutation will affect a specific tissue due to its specific needs, whether those may be high energy requirements or a need for the catabolism or anabolism of a specific neurotransmitter or nucleic acid. Because several copies of the mitochondrial genome are carried by each mitochondrion (2-10 in humans), mitochondrial mutations can be inherited maternally by mtDNA mutations which are present in mitochondria inside the oocyte
Oocyte
An oocyte, ovocyte, or rarely ocyte, is a female gametocyte or germ cell involved in reproduction. In other words, it is an immature ovum, or egg cell. An oocyte is produced in the ovary during female gametogenesis. The female germ cells produce a primordial germ cell which undergoes a mitotic...
before fertilization, or (as stated above) through mutations in the chromosomes.
In humans, the heavy strand of mtDNA carries 28 genes and the light strand of mtDNA carries only 9 genes. Eight of the 9 genes on the light strand code for mitochondrial tRNA molecules. Human mtDNA consists of 16,569 nucleotide pairs. The entire molecule is regulated by only one regulatory region which contains the origins of replication of both heavy and light strands. The entire human mitochondrial DNA molecule has been mapped. The rate of mutation
Mutation rate
In genetics, the mutation rate is the chance of a mutation occurring in an organism or gene in each generation...
in mtDNA is calculated to be about ten times greater than that of nuclear DNA, possibly due to a paucity of DNA repair mechanisms. This high mutation rate leads to a high variation between mitochondria, not only among different species but even within the same species. It is calculated that if two humans are chosen randomly and their mtDNA is tested, they will have an average of between fifty and seventy different nucleotides. This may not seem like much, but when compared to the total number of nucleotides of a human mitochondrial DNA molecule (16,569), as much as 0.42% of the mtDNA varies between two people.
Genetic code variants
The genetic codeGenetic code
The genetic code is the set of rules by which information encoded in genetic material is translated into proteins by living cells....
is, for the most part, universal, with few exceptions: mitochondrial genetics includes some of these. For most organisms the "stop codon
Stop codon
In the genetic code, a stop codon is a nucleotide triplet within messenger RNA that signals a termination of translation. Proteins are based on polypeptides, which are unique sequences of amino acids. Most codons in messenger RNA correspond to the addition of an amino acid to a growing polypeptide...
s" are “UAA”, “UAG”, and “UGA”. In vertebrate mitochondria “AGA” and “AGG” are also stop codons, but not “UGA”, which codes for tryptophan
Tryptophan
Tryptophan is one of the 20 standard amino acids, as well as an essential amino acid in the human diet. It is encoded in the standard genetic code as the codon UGG...
instead. “AUA” codes for isoleucine
Isoleucine
Isoleucine is an α-amino acid with the chemical formula HO2CCHCHCH2CH3. It is an essential amino acid, which means that humans cannot synthesize it, so it must be ingested. Its codons are AUU, AUC and AUA....
in most organisms but for methionine
Methionine
Methionine is an α-amino acid with the chemical formula HO2CCHCH2CH2SCH3. This essential amino acid is classified as nonpolar. This amino-acid is coded by the codon AUG, also known as the initiation codon, since it indicates mRNA's coding region where translation into protein...
in vertebrate mitochondrial mRNA.
There are many other variations among the codes used by other mitochondrial m/tRNA, which happened not to be harmful to their organisms, and which can be used as a tool (along with other mutations among the mtDNA/RNA of different species) to determine relative proximity of common ancestry of related species. (The more related two species are, the more mtDNA/RNA mutations will be the same in their mitochondrial genome).
Using these techniques, it is estimated that the first mitochondria arose around 1.5 billion years ago. A generally accepted hypothesis
Endosymbiotic theory
The endosymbiotic theory concerns the mitochondria, plastids , and possibly other organelles of eukaryotic cells. According to this theory, certain organelles originated as free-living bacteria that were taken inside another cell as endosymbionts...
is that mitochondria originated as an aerobic
Aerobic organism
An aerobic organism or aerobe is an organism that can survive and grow in an oxygenated environment.Faculitative anaerobes grow and survive in an oxygenated environment and so do aerotolerant anaerobes.-Glucose:...
prokaryote
Prokaryote
The prokaryotes are a group of organisms that lack a cell nucleus , or any other membrane-bound organelles. The organisms that have a cell nucleus are called eukaryotes. Most prokaryotes are unicellular, but a few such as myxobacteria have multicellular stages in their life cycles...
in a symbiotic relationship
Symbiosis
Symbiosis is close and often long-term interaction between different biological species. In 1877 Bennett used the word symbiosis to describe the mutualistic relationship in lichens...
within an anaerobic
Anaerobic organism
An anaerobic organism or anaerobe is any organism that does not require oxygen for growth. It could possibly react negatively and may even die if oxygen is present...
eukaryote
Eukaryote
A eukaryote is an organism whose cells contain complex structures enclosed within membranes. Eukaryotes may more formally be referred to as the taxon Eukarya or Eukaryota. The defining membrane-bound structure that sets eukaryotic cells apart from prokaryotic cells is the nucleus, or nuclear...
.
Inheritance patterns
Mitochondrial disease
Mitochondrial diseases are a group of disorders caused by dysfunctional mitochondria, the organelles that are the "powerhouses" of the cell. Mitochondria are found in every cell of the human body except red blood cells...
s (diseases due to malfunction of mitochondria) can be inherited both maternally and through chromosomal inheritance, the way in which they are passed on from generation to generation can vary greatly depending on the disease. Mitochondrial genetic mutations that occur in the nuclear DNA can occur in any of the chromosomes (depending on the species). Mutations inherited through the chromosomes can be autosomal dominant or recessive and can also be sex-linked dominant or recessive. Chromosomal inheritance follows normal Mendelian laws
Mendelian inheritance
Mendelian inheritance is a scientific description of how hereditary characteristics are passed from parent organisms to their offspring; it underlies much of genetics...
, despite the fact that the phenotype of the disease may be masked.
Because of the complex ways in which mitochondrial and nuclear DNA "communicate" and interact, even seemingly simple inheritance is hard to diagnose. A mutation in chromosomal DNA may change a protein that regulates (increases or decreases) the production of another certain protein in the mitochondria or the cytoplasm; this may lead to slight, if any, noticeable symptoms. On the other hand, some devastating mtDNA mutations are easy to diagnose because of their widespread damage to muscular, neural, and/or hepatic tissues (among other high-energy and metabolism-dependent tissues) and because they are present in the mother and all the offspring.
Mitochondrial genome mutations are passed on 100% of the time from mother to all her offspring. The number of affected mtDNA molecules inherited by a specific offspring can vary greatly because
- the mitochondria within the fertilized oocyte is what the new life will have to begin with (in terms of mtDNA),
- the number of affected mitochondriaHeteroplasmyHeteroplasmy is the presence of a mixture of more than one type of an organellar genome within a cell or individual...
varies from cell (in this case, the fertilized oocyte) to cell depending both on the number it inherited from its mother cell and environmental factors which may favor mutant or wildtype mitochondrial DNA, - the number of mtDNA moleculesThreshold expressionThreshold expression is a phenomenon in which phenotypic expression of a mitochondrial disease within an organ system occurs when the severity of the mutation, relative number of mutant mtDNA, and reliance of the organ system on oxidative phosphorylation combine in such a way that ATP production of...
in the mitochondria varies from around two to ten.
It is possible, even in twin births, for one baby to receive more than half mutant mtDNA molecules while the other twin may receive only a tiny fraction of mutant mtDNA molecules with respect to wildtype (depending on how the twins divide from each other and how many mutant mitochondria happen to be on each side of the division). In a few cases, some mitochondria or a mitochondrion from the sperm cell enters the oocyte but paternal mitochondria
Paternal mtDNA transmission
In genetics, paternal mtDNA transmission and paternal mtDNA inheritance refer to the incidence of mitochondrial DNA being passed from a father to his offspring. Paternal mtDNA inheritance is observed in a small proportion of species; in general, mtDNA is passed unchanged from a mother to her...
are actively decomposed.
Replication, repair, transcription, and translation
Mitochondrial replication is controlled by nuclear genes and is specifically suited to make as many mitochondria as that particular cell needs at the time. Human mitochondrial DNA (mtDNA) has three promoters, H1, H2, and L (heavy strand 1, heavy strand 2, and light strand promoters). The H2 promoter transcribes almost the entire heavy strand and the L promoter transcribes the entire light strand. The H1 promoter causes the transcription of the two mitochondrial rRNA molecules. When transcription takes place on the heavy strand a polycistronic transcript is created. The light strand produces either small transcripts, which can be used as primersPrimer (molecular biology)
A primer is a strand of nucleic acid that serves as a starting point for DNA synthesis. They are required for DNA replication because the enzymes that catalyze this process, DNA polymerases, can only add new nucleotides to an existing strand of DNA...
, or one long transcript. The production of primers occurs by processing of light strand transcripts with the Mitochondrial RNase MRP (Mitochondrial RNA Processing). The requirement of transcription to produce primers links the process of transcription to mtDNA replication. Full length transcripts are cut into functional tRNA, rRNA, and mRNA molecules. The process of transcription initiation in mitochondria involves three types of proteins: the mitochondrial RNA polymerase (POLRMT
POLRMT
DNA-directed RNA polymerase, mitochondrial is an enzyme that in humans is encoded by the POLRMT gene.- Function :This gene encodes a mitochondrial DNA-directed RNA polymerase. The gene product is responsible for mitochondrial gene expression as well as for providing RNA primers for initiation of...
), mitochondrial transcription factor A (TFAM), and mitochondrial transcription factors B1 and B2 (TFB1M, TFB2M). POLRMT
POLRMT
DNA-directed RNA polymerase, mitochondrial is an enzyme that in humans is encoded by the POLRMT gene.- Function :This gene encodes a mitochondrial DNA-directed RNA polymerase. The gene product is responsible for mitochondrial gene expression as well as for providing RNA primers for initiation of...
, TFAM, and TFB1M or TFB2M assemble at the mitochondrial promoters and begin transcription. The actual molecular events that are involved in initiation are unknown, but these factors make up the basal transcription machinery and have been shown to function in vitro. Mitochondrial translation is still not very well understood. In vitro
In vitro
In vitro refers to studies in experimental biology that are conducted using components of an organism that have been isolated from their usual biological context in order to permit a more detailed or more convenient analysis than can be done with whole organisms. Colloquially, these experiments...
translations have still not been successful, probably due to the difficulty of isolating sufficient mt mRNA, functional mt rRNA, and possibly because of the complicated changes that the mRNA undergoes before it is translated.
Mitochondrial DNA polymerase
The Mitochondrial DNA Polymerase (Pol gamma, encoded by the POLGPOLG
DNA polymerase subunit gamma is an enzyme that in humans is encoded by the POLG gene.-Functions:POLG is the gene that codes for the catalytic subunit of the mitochondrial DNA polymerase, called DNA polymerase gamma . The human POLG cDNA and gene were originally cloned mapped to chromosome 15,...
gene) is used in the copying of mtDNA during replication. Because the two (heavy
Heavy strand
Circular molecules of DNA, such as plasmids and typical mitochondrial genomes, consist of two strands of DNA called the heavy strand and the light strand . The two strands have different masses due to different proportions of heavier nucleic acids...
and light) strands on the circular mtDNA molecule have different origins of replication
Origin of replication
The origin of replication is a particular sequence in a genome at which replication is initiated. This can either be DNA replication in living organisms such as prokaryotes and eukaryotes, or RNA replication in RNA viruses, such as double-stranded RNA viruses...
, it replicates in a D-loop mode
D-loop replication
D-loop replication is a process by which chloroplasts and mitochondria replicate their genetic material. An important component of understanding D-loop replication is that many chloroplasts and mitochondria have a single circular chromosome like bacteria instead of the linear chromosomes found in...
. One strand begins to replicate first, displacing the other strand. This continues until replication reaches the origin of replication on the other strand, at which point the other strand begins replicating in the opposite direction. This results in two new mtDNA molecules. Each mitochondrion has several copies of the mtDNA molecule and the number of mtDNA molecules is a limiting factor in mitochondrial fission
Mitochondrial fission
Mitochondria can divide by fission and since they require mitochondrial DNA for their function, fission is coordinated with DNA replication. Some of the proteins that are involved in mitochondrial fission have been identified and some of them are associated with mitochondrial...
. After the mitochondrion has enough mtDNA, membrane area, and membrane proteins, it can undergo fission (very similar to that which bacteria use) to become two mitochondria. Evidence suggests that mitochondria can also undergo fusion and exchange (in a form of crossover
Chromosomal crossover
Chromosomal crossover is an exchange of genetic material between homologous chromosomes. It is one of the final phases of genetic recombination, which occurs during prophase I of meiosis in a process called synapsis. Synapsis begins before the synaptonemal complex develops, and is not completed...
) genetic material among each other. Mitochondria sometimes form large matrices in which fusion, fission, and protein exchanges are constantly occurring. mtDNA shared among mitochondria (despite the fact that they can undergo fusion).
Damage and transcription error
Mitochondrial DNA is susceptible to damage from free oxygen radicals from mistakes that occur during the production of ATP through the electron transport chain. These mistakes can be caused by genetic disorders, cancer, and temperature variations. These radicals can damage mtDNA molecules or change them, making it hard for mitochondrial polymerase to replicate them. Both cases can lead to deletions, rearrangements, and other mutations. Recent evidence has suggested that mitochondria have enzymes that proofread mtDNA and fix mutations that may occur due to free radicals. It is believed that a DNA recombinase found in mammalian cells is also involved in a repairing recombination process. Deletions and mutations due to free radicals have been associated with the aging process. It is believed that radicals cause mutations which lead to mutant proteins, which in turn lead to more radicals. This process takes many years and is associated with some aging processes involved in oxygen-dependent tissues such as brain, heart, muscle, and kidney. Auto-enhancing processes such as these are possible causes of degenerative diseases including Parkinson’sParkinson's disease
Parkinson's disease is a degenerative disorder of the central nervous system...
, Alzheimer’s
Alzheimer's disease
Alzheimer's disease also known in medical literature as Alzheimer disease is the most common form of dementia. There is no cure for the disease, which worsens as it progresses, and eventually leads to death...
, and coronary artery disease
Coronary heart disease
Coronary artery disease is the end result of the accumulation of atheromatous plaques within the walls of the coronary arteries that supply the myocardium with oxygen and nutrients. It is sometimes also called coronary heart disease...
.
Chromosomally mediated mtDNA replication errors
Because mitochondrial growth and fission are mediated by the nuclear DNA, mutations in nuclear DNA can have a wide array of effects on mtDNA replication. Despite the fact that the loci for some of these mutations have been found on human chromosomes, specific genes and proteins involved have not yet been isolated. Mitochondria need a certain protein to undergo fission. If this protein (made by the nucleus) is not present, the mitochondria grow but they do not divide. This leads to giant, inefficient mitochondria. Mistakes in chromosomal genes or their products can also affect mitochondrial replication more directly by inhibiting mitochondrial polymerase and can even cause mutations in the mtDNA directly and indirectly. Indirect mutations are most often caused by radicals created by defective proteins made from nuclear DNA.Mitochondrial diseases
Mitochondrial diseaseMitochondrial disease
Mitochondrial diseases are a group of disorders caused by dysfunctional mitochondria, the organelles that are the "powerhouses" of the cell. Mitochondria are found in every cell of the human body except red blood cells...
s range in severity from asymptomatic to fatal, and are most commonly due to inherited rather than acquired mutations of mitochondrial DNA. A given mitochondrial mutation can cause various diseases depending on the severity of the problem in the mitochondria and the tissue the affected mitochondria are in. Conversely, several different mutations may present themselves as the same disease. This almost patient-specific characterization of mitochondrial diseases (see Personalized medicine
Personalized medicine
Personalized medicine is a medical model emphasizing in general the customization of healthcare, with all decisions and practices being tailored to individual patients in whatever ways possible...
) makes them very hard to accurately recognize, diagnose and trace. Some diseases are observable at or even before birth (many causing death) while others do not show themselves until late adulthood (late-onset disorders). This is because the number of mutant versus wildtype mitochondria varies between cells and tissues, and is continuously changing. Because cells have multiple mitochondria, different mitochondria in the same cell can have different variations of the mtDNA. This condition is referred to as heteroplasmy
Heteroplasmy
Heteroplasmy is the presence of a mixture of more than one type of an organellar genome within a cell or individual...
. When a certain tissue reaches a certain ratio of mutant versus wildtype mitochondria, a disease will present itself. The ratio varies from person to person and tissue to tissue (depending on its specific energy, oxygen, and metabolism requirements, and the effects of the specific mutation). Mitochondrial diseases are very numerous and different. Apart from diseases caused by abnormalities in mitochondrial DNA, many diseases are suspected to be associated in part by mitochondrial dysfunctions, such as diabetes mellitus
Diabetes mellitus
Diabetes mellitus, often simply referred to as diabetes, is a group of metabolic diseases in which a person has high blood sugar, either because the body does not produce enough insulin, or because cells do not respond to the insulin that is produced...
, forms of cancer
Cancer
Cancer , known medically as a malignant neoplasm, is a large group of different diseases, all involving unregulated cell growth. In cancer, cells divide and grow uncontrollably, forming malignant tumors, and invade nearby parts of the body. The cancer may also spread to more distant parts of the...
and cardiovascular disease
Cardiovascular disease
Heart disease or cardiovascular disease are the class of diseases that involve the heart or blood vessels . While the term technically refers to any disease that affects the cardiovascular system , it is usually used to refer to those related to atherosclerosis...
, lactic acidosis
Lactic acidosis
Lactic acidosis is a physiological condition characterized by low pH in body tissues and blood accompanied by the buildup of lactate especially D-lactate, and is considered a distinct form of metabolic acidosis. The condition typically occurs when cells receive too little oxygen , for example...
, specific forms of myopathy
Myopathy
In medicine, a myopathy is a muscular disease in which the muscle fibers do not function for any one of many reasons, resulting in muscular weakness. "Myopathy" simply means muscle disease...
, osteoporosis
Osteoporosis
Osteoporosis is a disease of bones that leads to an increased risk of fracture. In osteoporosis the bone mineral density is reduced, bone microarchitecture is deteriorating, and the amount and variety of proteins in bone is altered...
, Alzheimer's disease
Alzheimer's disease
Alzheimer's disease also known in medical literature as Alzheimer disease is the most common form of dementia. There is no cure for the disease, which worsens as it progresses, and eventually leads to death...
, Parkinsons's disease, stroke
Stroke
A stroke, previously known medically as a cerebrovascular accident , is the rapidly developing loss of brain function due to disturbance in the blood supply to the brain. This can be due to ischemia caused by blockage , or a hemorrhage...
, Male infertility
Male infertility
Male infertility refers to the inability of a male to achieve a pregnancy in a fertile female. In humans it accounts for 40-50% of infertility. Male infertility is commonly due to deficiencies in the semen, and semen quality is used as a surrogate measure of male fecundity.-Pre-testicular...
and which are also believed to play a role in the aging process.
See also
- Paternal mtDNA transmissionPaternal mtDNA transmissionIn genetics, paternal mtDNA transmission and paternal mtDNA inheritance refer to the incidence of mitochondrial DNA being passed from a father to his offspring. Paternal mtDNA inheritance is observed in a small proportion of species; in general, mtDNA is passed unchanged from a mother to her...
- Human mitochondrial DNA haplogroupsHuman mitochondrial DNA haplogroupsIn human genetics, a human mitochondrial DNA haplogroup is a haplogroup defined by differences in human mitochondrial DNA. Haplogroups are used to represent the major branch points on the mitochondrial phylogenetic tree...
- Cambridge Reference SequenceCambridge Reference SequenceThe Cambridge Reference Sequence for human mitochondrial DNA was first published in 1981 leading to the initiation of the human genome project.A group under Dr...
- Human mitochondrial molecular clockHuman mitochondrial molecular clockThe human mitochondrial molecular clock is the rate at which mutations have been accumulating in the mitochondrial genome of hominids during the course of human evolution. The archeological record of human activity from early periods in human prehistory is relatively limited and its interpretation...
- Genetic genealogy for lists of databases which help users find others with their Y-DNA and mtDNA.