

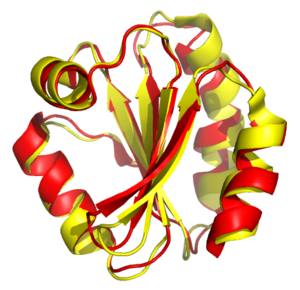
Structural alignment
Encyclopedia
Polymer
A polymer is a large molecule composed of repeating structural units. These subunits are typically connected by covalent chemical bonds...
structures based on their shape and three-dimensional conformation
Tertiary structure
In biochemistry and molecular biology, the tertiary structure of a protein or any other macromolecule is its three-dimensional structure, as defined by the atomic coordinates.-Relationship to primary structure:...
. This process is usually applied to protein
Protein
Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form, facilitating a biological function. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of...
tertiary structure
Tertiary structure
In biochemistry and molecular biology, the tertiary structure of a protein or any other macromolecule is its three-dimensional structure, as defined by the atomic coordinates.-Relationship to primary structure:...
s but can also be used for large RNA
RNA
Ribonucleic acid , or RNA, is one of the three major macromolecules that are essential for all known forms of life....
molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
techniques. Structural alignment can therefore be used to imply evolution
Evolution
Evolution is any change across successive generations in the heritable characteristics of biological populations. Evolutionary processes give rise to diversity at every level of biological organisation, including species, individual organisms and molecules such as DNA and proteins.Life on Earth...
ary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution
Convergent evolution
Convergent evolution describes the acquisition of the same biological trait in unrelated lineages.The wing is a classic example of convergent evolution in action. Although their last common ancestor did not have wings, both birds and bats do, and are capable of powered flight. The wings are...
by which multiple unrelated amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
sequences converge on a common tertiary structure
Tertiary structure
In biochemistry and molecular biology, the tertiary structure of a protein or any other macromolecule is its three-dimensional structure, as defined by the atomic coordinates.-Relationship to primary structure:...
.
Structural alignments can compare two sequences or multiple sequences
Multiple sequence alignment
A multiple sequence alignment is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a lineage and are descended from a common ancestor...
. Because these alignments rely on information about all the query sequences' three-dimensional conformations, the method can only be used on sequences where these structures are known. These are usually found by X-ray crystallography
X-ray crystallography
X-ray crystallography is a method of determining the arrangement of atoms within a crystal, in which a beam of X-rays strikes a crystal and causes the beam of light to spread into many specific directions. From the angles and intensities of these diffracted beams, a crystallographer can produce a...
or NMR spectroscopy
NMR spectroscopy
Nuclear magnetic resonance spectroscopy, most commonly known as NMR spectroscopy, is a research technique that exploits the magnetic properties of certain atomic nuclei to determine physical and chemical properties of atoms or the molecules in which they are contained...
. It is possible to perform a structural alignment on structures produced by structure prediction
Protein structure prediction
Protein structure prediction is the prediction of the three-dimensional structure of a protein from its amino acid sequence — that is, the prediction of its secondary, tertiary, and quaternary structure from its primary structure. Structure prediction is fundamentally different from the inverse...
methods. Indeed, evaluating such predictions often requires a structural alignment between the model and the true known structure to assess the model's quality. Structural alignments are especially useful in analyzing data from structural genomics
Structural genomics
Structural genomics seeks to describe the 3-dimensional structure of every protein encoded by a given genome. This genome-based approach allows for a high-throughput method of structure determination by a combination of experimental and modeling approaches...
and proteomics
Proteomics
Proteomics is the large-scale study of proteins, particularly their structures and functions. Proteins are vital parts of living organisms, as they are the main components of the physiological metabolic pathways of cells. The term "proteomics" was first coined in 1997 to make an analogy with...
efforts, and they can be used as comparison points to evaluate alignments produced by purely sequence-based bioinformatics
Bioinformatics
Bioinformatics is the application of computer science and information technology to the field of biology and medicine. Bioinformatics deals with algorithms, databases and information systems, web technologies, artificial intelligence and soft computing, information and computation theory, software...
methods.
The outputs of a structural alignment are a superposition of the atomic coordinate sets and a minimal root mean square
Root mean square
In mathematics, the root mean square , also known as the quadratic mean, is a statistical measure of the magnitude of a varying quantity. It is especially useful when variates are positive and negative, e.g., sinusoids...
deviation (RMSD) between the structures. The RMSD of two aligned structures indicates their divergence from one another. Structural alignment can be complicated by the existence of multiple protein domain
Protein domain
A protein domain is a part of protein sequence and structure that can evolve, function, and exist independently of the rest of the protein chain. Each domain forms a compact three-dimensional structure and often can be independently stable and folded. Many proteins consist of several structural...
s within one or more of the input structures, because changes in relative orientation of the domains between two structures to be aligned can artificially inflate the RMSD.
Data produced by structural alignment
The minimum information produced from a successful structural alignment is a set of superposed three-dimensional coordinates for each input structure. (Note that one input element may be fixed as a reference and therefore its superposed coordinates do not change.) The fitted structures can be used to calculate mutual RMSD values, as well as other more sophisticated measures of structural similarity such as the global distance testGlobal distance test
The global distance test or GDT is a measure of similarity between two protein structures with identical amino acid sequences but different tertiary structures...
(GDT, the metric used in CASP
CASP
CASP, which stands for Critical Assessment of Techniques for Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994...
). The structural alignment also implies a corresponding one-dimensional sequence alignment
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
from which a sequence identity, or the percentage of residues that are identical between the input structures, can be calculated as a measure of how closely the two sequences are related.
Types of comparisons
Because protein structures are composed of amino acidAmino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
s whose side chain
Side chain
In organic chemistry and biochemistry, a side chain is a chemical group that is attached to a core part of the molecule called "main chain" or backbone. The placeholder R is often used as a generic placeholder for alkyl group side chains in chemical structure diagrams. To indicate other non-carbon...
s are linked by a common protein backbone, a number of different possible subsets of the atoms that make up a protein macromolecule can be used in producing a structural alignment and calculating the corresponding RMSD values. When aligning structures with very different sequences, the side chain atoms generally are not taken into account because their identities differ between many aligned residues. For this reason it is common for structural alignment methods to use by default only the backbone atoms included in the peptide bond
Peptide bond
This article is about the peptide link found within biological molecules, such as proteins. A similar article for synthetic molecules is being created...
. For simplicity and efficiency, often only the alpha carbon
Alpha carbon
The alpha carbon in organic chemistry refers to the first carbon that attaches to a functional group . By extension, the second carbon is the beta carbon, and so on....
positions are considered, since the peptide bond has a minimally variant planar conformation. Only when the structures to be aligned are highly similar or even identical is it meaningful to align side-chain atom positions, in which case the RMSD reflects not only the conformation of the protein backbone but also the rotameric states of the side chains. Other comparison criteria that reduce noise and bolster positive matches include secondary structure
Secondary structure
In biochemistry and structural biology, secondary structure is the general three-dimensional form of local segments of biopolymers such as proteins and nucleic acids...
assignment, native contact
Native contact
In protein folding, a native contact is a contact between the side chains of two amino acids that are not neighboring in the amino acid sequence but are spatially close in the protein's native state tertiary structure...
maps or residue interaction patterns, measures of side chain packing, and measures of hydrogen bond
Hydrogen bond
A hydrogen bond is the attractive interaction of a hydrogen atom with an electronegative atom, such as nitrogen, oxygen or fluorine, that comes from another molecule or chemical group. The hydrogen must be covalently bonded to another electronegative atom to create the bond...
retention.
Structural superposition
The most basic possible comparison between protein structures makes no attempt to align the input structures and requires a precalculated alignment as input to determine which of the residues in the sequence are intended to be considered in the RMSD calculation. Structural superposition is commonly used to compare multiple conformations of the same protein (in which case no alignment is necessary, since the sequences are the same) and to evaluate the quality of alignments produced using only sequence information between two or more sequences whose structures are known. This method traditionally uses a simple least-squares fitting algorithm, in which the optimal rotations and translations are found by minimizing the sum of the squared distances among all structures in the superposition. More recently, maximum likelihood and Bayesian methods have greatly increased the accuracy of the estimated rotations, translations, and covariance matrices for the superposition.Algorithms based on multidimensional rotations and modified quaternion
Quaternion
In mathematics, the quaternions are a number system that extends the complex numbers. They were first described by Irish mathematician Sir William Rowan Hamilton in 1843 and applied to mechanics in three-dimensional space...
s have been developed to identify topological relationships between protein structures without the need for a predetermined alignment. Such algorithms have successfully identified canonical folds such as the four-helix bundle
Helix bundle
A helix bundle is a small protein fold composed of several alpha helices that are usually nearly parallel or antiparallel to each other.-Three-helix bundles:Three-helix bundles are among the smallest and fastest known cooperatively folding structural domains...
. The SuperPose method is sufficiently extensible to correct for relative domain rotations and other structural pitfalls.
Optimal solution
the optimal "threading" of a protein sequence onto a known structure and the production of an optimal multiple sequence alignment have been shown to be NP-completeNP-complete
In computational complexity theory, the complexity class NP-complete is a class of decision problems. A decision problem L is NP-complete if it is in the set of NP problems so that any given solution to the decision problem can be verified in polynomial time, and also in the set of NP-hard...
. However, this does not imply that the structural alignment problem is NP-complete. Strictly speaking, an optimal solution to the protein structure alignment problem is only known for certain protein structure similarity measures, such as the measures used in protein structure prediction experiments, GDT_TS and MaxSub. These measures can be rigorously optimized using an algorithm capable of maximizing the number of atoms in two proteins that can be superimposed under a predefined distance cutoff. Unfortunately, the algorithm for optimal solution is not practical, since its running time depends not only on the lengths but also on the intrinsic geometry of input proteins.
Approximate solution
Approximate polynomial-time algorithms for structural alignment that produce a family of "optimal" solutions within an approximation parameter for a given scoring function have been developed. Although these algorithms theoretically classify the approximate protein structure alignment problem as "tractable", they are still computationally too expensive for large scale protein structure analysis. As a consequence, practical algorithms that converge to the global solutions of the alignment, given a scoring function, do not exist. Most algorithms are, therefore, heuristic, but algorithms that guarantee the convergence to at least local maximizers of the scoring functions, and are practical, have been developed.Representation of structures
Protein structures have to be represented in some coordinate-independent space to make them comparable. This is typically achieved by constructing a sequence-to-sequence matrix or series of matrices that encompass comparative metrics: rather than absolute distances relative to a fixed coordinate space. An intuitive representation is the distance matrixDistance matrix
In mathematics, computer science and graph theory, a distance matrix is a matrix containing the distances, taken pairwise, of a set of points...
, which is a two-dimensional matrix
Matrix (mathematics)
In mathematics, a matrix is a rectangular array of numbers, symbols, or expressions. The individual items in a matrix are called its elements or entries. An example of a matrix with six elements isMatrices of the same size can be added or subtracted element by element...
containing all pairwise distances between some subset of the atoms in each structure (such as the alpha carbon
Alpha carbon
The alpha carbon in organic chemistry refers to the first carbon that attaches to a functional group . By extension, the second carbon is the beta carbon, and so on....
s). The matrix increases in dimensionality as the number of structures to be simultaneously aligned increases. Reducing the protein to a coarse metric such as secondary structure
Secondary structure
In biochemistry and structural biology, secondary structure is the general three-dimensional form of local segments of biopolymers such as proteins and nucleic acids...
elements (SSEs) or structural fragments can also produce sensible alignments, despite the loss of information from discarding distances, as noise is also discarded. Choosing a representation to facilitate computation is critical to developing an efficient alignment mechanism.
Methods
Structural alignment techniques have been used in comparing individual structures or sets of structures as well as in the production of "all-to-all" comparison databases that measure the divergence between every pair of structures present in the Protein Data BankProtein Data Bank
The Protein Data Bank is a repository for the 3-D structural data of large biological molecules, such as proteins and nucleic acids....
(PDB). Such databases are used to classify proteins by their fold
Tertiary structure
In biochemistry and molecular biology, the tertiary structure of a protein or any other macromolecule is its three-dimensional structure, as defined by the atomic coordinates.-Relationship to primary structure:...
.
DALI
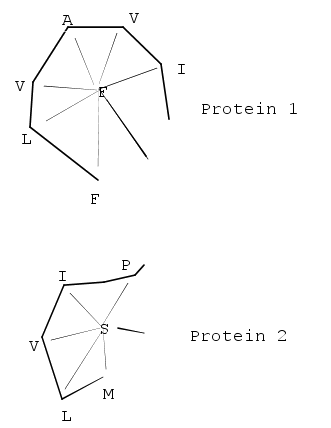
Secondary structure
In biochemistry and structural biology, secondary structure is the general three-dimensional form of local segments of biopolymers such as proteins and nucleic acids...
features that involve residues that are contiguous in sequence appear on the matrix's main diagonal; other diagonals in the matrix reflect spatial contacts between residues that are not near each other in the sequence. When these diagonals are parallel to the main diagonal, the features they represent are parallel; when they are perpendicular, their features are antiparallel. This representation is memory-intensive because the features in the square matrix are symmetrical (and thus redundant) about the main diagonal.
When two proteins' distance matrices share the same or similar features in approximately the same positions, they can be said to have similar folds with similar-length loops connecting their secondary structure elements. DALI's actual alignment process requires a similarity search after the two proteins' distance matrices are built; this is normally conducted via a series of overlapping submatrices of size 6x6. Submatrix matches are then reassembled into a final alignment via a standard score-maximization algorithm - the original version of DALI used a Monte Carlo
Monte Carlo method
Monte Carlo methods are a class of computational algorithms that rely on repeated random sampling to compute their results. Monte Carlo methods are often used in computer simulations of physical and mathematical systems...
simulation to maximize a structural similarity score that is a function of the distances between putative corresponding atoms. In particular, more distant atoms within corresponding features are exponentially downweighted to reduce the effects of noise introduced by loop mobility, helix torsions, and other minor structural variations. Because DALI relies on an all-to-all distance matrix, it can account for the possibility that structurally aligned features might appear in different orders within the two sequences being compared.
The DALI method has also been used to construct a database known as FSSP
Families of structurally similar proteins
Families of Structurally Similar Proteins or FSSP is a database of structurally superimposed proteins generated using the "Distance-matrix ALIgnment" algorithm. The database is helpful for the comparison of protein structures.-External links:*...
(Fold classification based on Structure-Structure alignment of Proteins, or Families of Structurally Similar Proteins) in which all known protein structures are aligned with each other to determine their structural neighbors and fold classification. There is an searchable database based on DALI as well as a downloadable program and web search based on a standalone version known as DaliLite.
Combinatorial extension
The combinatorial extension (CE) method is similar to DALI in that it too breaks each structure in the query set into a series of fragments that it then attempts to reassemble into a complete alignment. A series of pairwise combinations of fragments called aligned fragment pairs, or AFPs, are used to define a similarity matrix through which an optimal path is generated to identify the final alignment. Only AFPs that meet given criteria for local similarity are included in the matrix as a means of reducing the necessary search space and thereby increasing efficiency. A number of similarity metrics are possible; the original definition of the CE method included only structural superpositions and inter-residue distances but has since been expanded to include local environmental properties such as secondary structure, solvent exposure, hydrogen-bonding patterns, and dihedral angleDihedral angle
In geometry, a dihedral or torsion angle is the angle between two planes.The dihedral angle of two planes can be seen by looking at the planes "edge on", i.e., along their line of intersection...
s.
An alignment path is calculated as the optimal path through the similarity matrix by linearly progressing through the sequences and extending the alignment with the next possible high-scoring AFP pair. The initial AFP pair that nucleates the alignment can occur at any point in the sequence matrix. Extensions then proceed with the next AFP that meets given distance criteria restricting the alignment to low gap sizes. The size of each AFP and the maximum gap size are required input parameters but are usually set to empirically determined values of 8 and 30 respectively. Like DALI and SSAP, CE has been used to construct an all-to-all fold classification database from the known protein structures in the PDB.
The RCSB PDB
Protein Data Bank
The Protein Data Bank is a repository for the 3-D structural data of large biological molecules, such as proteins and nucleic acids....
has recently released an updated version of CE and FATCAT as part of the RCSB PDB Protein Comparison Tool. It provides a new variation of CE that can detect circular permutations in protein structures.
GANGSTA+
GANGSTA+ is a combinatorial algorithm for non-sequential structural alignment of proteins and similarity search in databases (http://gangsta.chemie.fu-berlin.de).It uses a combinatorial approach on the secondary structure level to evaluate similarities between two protein structures based on contact maps. Different SSE assignment modes can be used. The assignment of SSEs can be performed respecting the sequential order of the SSEs in the polypeptide chains of the considered protein pair (sequential alignment) or by ignoring this order (non-sequential alignment). Furthermore, SSE pairs can optionally be aligned in reverse orientation.
The highest ranking SSE assignments are transferred to the residue level by a pointmatching approach. To obtain an initial common set of atomic coordinates for both proteins, pairwise attractive interactions of the C-alpha atom pairs are defined by inverse Lorentzians and energy minimized.
MAMMOTH
MAtching Molecular Models Obtained from THeory. As its name suggests, MAMMOTH was originally developed for comparing models coming from structure prediction (THeory) since it is tolerant of large unalignable regions, but it has proven to work well with experimental models, especially when looking for remote homology. Benchmarks on targets of blind structure prediction (the CASPCASP
CASP, which stands for Critical Assessment of Techniques for Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994...
experiment) and automated GO annotation have shown it is tightly rank correlated with human curated annotation. A highly complete database of mammoth-based structure annotations for the predicted structures of unknown proteins covering 150 genomes facilitates genomic scale normalization.
MAMMOTH-based structure alignment methods decompose the protein structure into short, seven-residue, peptides (heptapeptides) which are compared with the heptapeptides of another protein. The similarity score between two heptapeptides is calculated using a unit-vector RMS (URMS) method. These scores are stored in a similarity matrix, and with a hybrid (local-global) dynamic programming
Dynamic programming
In mathematics and computer science, dynamic programming is a method for solving complex problems by breaking them down into simpler subproblems. It is applicable to problems exhibiting the properties of overlapping subproblems which are only slightly smaller and optimal substructure...
the optimal residue alignment is calculated. Protein similarity scores calculated with MAMMOTH is derived from the likelihood of obtaining a given structural alignment by chance.
MAMMOTH-mult is an extension of the MAMMOTH algorithm to be used to align related families of protein structures. This algorithm is very fast and produces consistent and high quality structural alignments. Multiple structural alignments calculated with MAMMOTH-mult produces structurally-implied sequence alignments that can be further used for multiple-template homology modeling, HMM
Hidden Markov model
A hidden Markov model is a statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved states. An HMM can be considered as the simplest dynamic Bayesian network. The mathematics behind the HMM was developed by L. E...
-based protein structure prediction
Protein structure prediction
Protein structure prediction is the prediction of the three-dimensional structure of a protein from its amino acid sequence — that is, the prediction of its secondary, tertiary, and quaternary structure from its primary structure. Structure prediction is fundamentally different from the inverse...
, and profile-type PSI-BLAST searches.
RAPIDO
Rapid Alignment of Proteins In terms of DOmains. RAPIDO is a web server for the 3D alignment of crystal structures of different protein molecules, in the presence of conformational changes. Similar to what CE does as a first step, RAPIDO identifies fragments that are structurally similar in the two proteins using an approach based on difference distance matrices. The Matching Fragment Pairs (MFPs) are then represented as nodes in a graph which are chained together to form an alignment by means of an algorithm for the identification of the longest path on a DAG (Directed Acyclic Graph). The final step of refinement is performed to improve the quality of the alignment. After aligning the two structures the server applies a genetic algorithm for the identification of conformationally invariant regions. These regions correspond to groups of atoms whose interatomic distances are constant (within a defined tolerance). In doing so RAPIDO takes into account the variation in the reliability of atomic coordinates by employing weighting-functions based on the refined B-values. The regions identified as conformationally invariant by RAPIDO represent reliable sets of atoms for the superposition of the two structures that can be used for a detailed analysis of changes in the conformation. In addition to the functionalities provided by existing tools, RAPIDO can identify structurally equivalent regions even when these consist of fragments that are distant in terms of sequence and separated by other movable domains.SABERTOOTH
SABERTOOTH uses structural profiles to perform structural alignments. The underlying structural profiles expresses the global connectivity of each residue. Despite the very condensed vectorial representation, the tool recognizes structural similarities with accuracy comparable to established alignment tools based on coordinates and performs comparably in quality. Furthermore, the algorithm has favourable scaling of computation time with chain length. Since the algorithm is independent of the details of the structural representation, the framework can be generalized to sequence-to-sequence and sequence-to-structure comparison within the same setup, and it is therefore more general than other tools.SABERTOOTH can be used online at http://www.fkp.tu-darmstadt.de/sabertooth/
SSAP
The SSAP (Sequential Structure Alignment Program) method uses double dynamic programmingDynamic programming
In mathematics and computer science, dynamic programming is a method for solving complex problems by breaking them down into simpler subproblems. It is applicable to problems exhibiting the properties of overlapping subproblems which are only slightly smaller and optimal substructure...
to produce a structural alignment based on atom-to-atom vectors in structure space. Instead of the alpha carbons typically used in structural alignment, SSAP constructs its vectors from the beta carbons for all residues except glycine, a method which thus takes into account the rotameric state of each residue as well as its location along the backbone. SSAP works by first constructing a series of inter-residue distance vectors between each residue and its nearest non-contiguous neighbors on each protein. A series of matrices are then constructed containing the vector differences between neighbors for each pair of residues for which vectors were constructed. Dynamic programming applied to each resulting matrix determines a series of optimal local alignments which are then summed into a "summary" matrix to which dynamic programming is applied again to determine the overall structural alignment.
SSAP originally produced only pairwise alignments but has since been extended to multiple alignments as well. It has been applied in an all-to-all fashion to produce a hierarchical fold classification scheme known as CATH
CATH
The CATH Protein Structure Classification is a semi-automatic, hierarchical classification of protein domains published in 1997 by Christine Orengo, Janet Thornton and their colleagues....
(Class, Architecture, Topology, Homology), which has been used to construct the CATH Protein Structure Classification database.
TOPOFIT
In the TOPOFIT method, of protein structures is analyzed using three-dimensional Delaunay triangulation patterns derived from backbone representation. It has been found that structurally related proteins have a common spatial invariant part, a set of tetrahedrons, mathematically described as a common spatial sub-graph volume of the three-dimensional contact graph derived from Delaunay tessellation (DT). Based on this property of protein structures we present a novel common volume superimposition (TOPOFIT) method to produce structural alignments of proteins. The superimposition of the DT patterns allows one to objectively identify a common number of equivalent residues in the structural alignment, in other words, TOPOFIT identifies a feature point on the RMSD/Ne curve, a topomax point, until which two structures correspond to each other including backbone and inter-residue contacts, while the growing number of mismatches between the DT patterns occurs at larger RMSD (Ne) after topomax point. The topomax point is present in all alignments from different protein structural classes; therefore, the TOPOFIT method identifies common, invariant structural parts between proteins. The TOPOFIT method adds new opportunities for the comparative analysis of protein structures and for more detailed studies on understanding the molecular principles of tertiary structure organization and functionality. It helps to detect conformational changes, topological differences in variable parts, which are particularly important for studies of variations in active/binding sites and protein classification.SSM
Secondary Structure Matching (SSM), or PDBeFold at the Protein Data Bank in Europe uses graph matching followed by c-alpha alignment to compute alignments.Recent developments
Improvements in structural alignment methods constitute an active area of research, and new or modified methods are often proposed that are claimed to offer advantages over the older and more widely distributed techniques. A recent example, TM-align, uses a novel method for weighting its distance matrix, to which standard dynamic programmingDynamic programming
In mathematics and computer science, dynamic programming is a method for solving complex problems by breaking them down into simpler subproblems. It is applicable to problems exhibiting the properties of overlapping subproblems which are only slightly smaller and optimal substructure...
is then applied. The weighting is proposed to accelerate the convergence of dynamic programming and correct for effects arising from alignment lengths. In a benchmarking study, TM-align has been reported to improve in both speed and accuracy over DALI and CE.
However, as algorithmic improvements and computer performance have erased purely technical deficiencies in older approaches, it has become clear that there is no one universal criterion for the 'optimal' structural alignment. TM-align, for instance, is particularly robust in quantifying comparisons between sets of proteins with great disparities in sequence lengths, but it only indirectly captures hydrogen bonding or secondary structure order conservation which might be better metrics for alignment of evolutionarily related proteins. Thus recent developments have focused on optimizing particular attributes such as speed, quantification of scores, correlation to alternative gold standards, or tolerance of imperfection in structural data or ab initio structural models. An alternative methodology that is gaining popularity is to use the consensus of various methods to ascertain proteins structural similarities.
In choosing among modern algorithms, investigators should strongly consider the optimization for the purpose of intended application, as well as on the algorithm's penetration into the particular field so as to facilitate comparison to other authors' results. For example, MAMMOTH has been specialized for speed and correlation to human annotation, and is thus suited for large-scale structural genomics studies: MAMMOTH has been adopted by Rosetta@home
Rosetta@home
Rosetta@home is a distributed computing project for protein structure prediction on the Berkeley Open Infrastructure for Network Computing platform, run by the Baker laboratory at the University of Washington...
and the World Community Grid
World Community Grid
World Community Grid is an effort to create the world's largest public computing grid to tackle scientific research projects that benefit humanity...
's Yeast
and Human Proteome Folding projects because it is designed for remote structural homology detection even with relatively inaccurate or incomplete predicted structure models. The venerable DALI
Families of structurally similar proteins
Families of Structurally Similar Proteins or FSSP is a database of structurally superimposed proteins generated using the "Distance-matrix ALIgnment" algorithm. The database is helpful for the comparison of protein structures.-External links:*...
is perhaps the most ubiquitous in the literature and due to its integration with other European Bioinformatics Institute
European Bioinformatics Institute
The European Bioinformatics Institute is a centre for research and services in bioinformatics, and is part of European Molecular Biology Laboratory...
web-based tools, the EBI DALI is easily approached by researchers interested in singleton applications.
Historically, it was initially unclear if comparisons that preserved sequence order would be more sensitive than ones that simply compare architecture or contacts without regard to secondary segment ordering. Early versions of DALI allowed a choice of non-sequential alignment at a great cost in speed. Non-sequential methods lost favor as segment order preserving methods outperformed them in speed, quantification of high-confidence similarity scores, and amenability to adoption of rich scoring heuristics. However, in some applications, discovery of conserved but out-of-order structure motif recognition is vital and additionally some forms of experimental data collection, such as cryo-electron microscopy, generally resolve regular secondary elements but not their connection order. This has renewed interest in Non-sequential approaches. Some examples are GANGSTA+ and TOPOFIT.
RNA structural alignment
Structural alignment techniques have traditionally been applied exclusively to proteins, as the primary biological macromoleculeMacromolecule
A macromolecule is a very large molecule commonly created by some form of polymerization. In biochemistry, the term is applied to the four conventional biopolymers , as well as non-polymeric molecules with large molecular mass such as macrocycles...
s that assume characteristic three-dimensional structures. However, large RNA
RNA
Ribonucleic acid , or RNA, is one of the three major macromolecules that are essential for all known forms of life....
molecules also form characteristic tertiary structures
RNA structure
Biomolecular structure is the structure of biomolecules, mainly proteins and the nucleic acids DNA and RNA. The structure of these molecules is frequently decomposed into primary structure, secondary structure, tertiary structure, and quaternary structure. The scaffold for this structure is...
, which are mediated primarily by hydrogen bond
Hydrogen bond
A hydrogen bond is the attractive interaction of a hydrogen atom with an electronegative atom, such as nitrogen, oxygen or fluorine, that comes from another molecule or chemical group. The hydrogen must be covalently bonded to another electronegative atom to create the bond...
s formed between base pair
Base pair
In molecular biology and genetics, the linking between two nitrogenous bases on opposite complementary DNA or certain types of RNA strands that are connected via hydrogen bonds is called a base pair...
s as well as base stacking. Functionally similar noncoding RNA molecules can be especially difficult to extract from genomics
Genomics
Genomics is a discipline in genetics concerning the study of the genomes of organisms. The field includes intensive efforts to determine the entire DNA sequence of organisms and fine-scale genetic mapping efforts. The field also includes studies of intragenomic phenomena such as heterosis,...
data because structure is more strongly conserved than sequence in RNA as well as in proteins, and the more limited alphabet of RNA decreases the information content
Information content
The term information content is used to refer the meaning of information as opposed to the form or carrier of the information. For example, the meaning that is conveyed in an expression or document, which can be distinguished from the sounds or symbols or codes and carrier that physically form the...
of any given nucleotide
Nucleotide
Nucleotides are molecules that, when joined together, make up the structural units of RNA and DNA. In addition, nucleotides participate in cellular signaling , and are incorporated into important cofactors of enzymatic reactions...
at any given position.
A recent method for pairwise structural alignment of RNA sequences with low sequence identity has been published and implemented in the program FOLDALIGN. However, this method is not truly analogous to protein structural alignment techniques because it computationally predicts the structures of the RNA input sequences rather than requiring experimentally determined structures as input. Although computational prediction of the protein folding
Protein folding
Protein folding is the process by which a protein structure assumes its functional shape or conformation. It is the physical process by which a polypeptide folds into its characteristic and functional three-dimensional structure from random coil....
process has not been particularly successful to date, RNA structures without pseudoknot
Pseudoknot
A pseudoknot is a nucleic acid secondary structure containing at least two stem-loop structures in which half of one stem is intercalated between the two halves of another stem. The pseudoknot was first recognized in the turnip yellow mosaic virus in 1982...
s can often be sensibly predicted using free energy
Thermodynamic free energy
The thermodynamic free energy is the amount of work that a thermodynamic system can perform. The concept is useful in the thermodynamics of chemical or thermal processes in engineering and science. The free energy is the internal energy of a system less the amount of energy that cannot be used to...
-based scoring methods that account for base pairing and stacking.
Software
Choosing a software tool for structural alignment can be a challenge due to the large variety of available packages that differ significantly in methodology and reliability. A partial solution to this problem was presented in and made publicly accessible through the ProCKSI webserver. A more complete list of currently available and freely distributed structural alignment software can be found in structural alignment softwareStructural alignment software
This list of structural comparison and alignment software is a compilation of software tools and web portals used in pairwise or multiple structural comparison and structural alignment.-Structural comparison and alignment:Key map:* Class:...
.
Properties of some structural alignment servers and software packages are summarized and tested with examples at Structural Alignment Tools in Proteopedia.Org.
See also
- Multiple sequence alignmentMultiple sequence alignmentA multiple sequence alignment is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a lineage and are descended from a common ancestor...
- List of sequence alignment software
- Sequence alignmentSequence alignmentIn bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are...
- Structural Classification of ProteinsStructural Classification of ProteinsThe Structural Classification of Proteins database is a largely manual classification of protein structural domains based on similarities of their structures and amino acid sequences. A motivation for this classification is to determine the evolutionary relationship between proteins...
Further reading
- Bourne PE, Shindyalov IN. (2003): Structure Comparison and Alignment. In: Bourne, P.E., Weissig, H. (Eds): Structural Bioinformatics. Hoboken NJ: Wiley-Liss. ISBN 0-471-20200-2
- Yuan X, Bystroff C.(2004) "Non-sequential Structure-based Alignments Reveal Topology-independent Core Packing Arrangements in Proteins", Bioinformatics. Nov 5, 2004
- Jung J, Lee B. (2000). Protein structure alignment using environmental profiles. Protein Eng 13:535-543.
- Ye Y, Godzik A. (2005). Multiple flexible structure alignment using partial order graphs Bioinformatics 21(10): 2362-2369 Abstract
- Sippl M, Wiederstein M (2008). A note on difficult structure alignment problems. Bioinformatics 24(3): 426-427 Full Text